Science|DrugCLIP:走向基因组级别的小分子筛选
今天给大家介绍一篇清华大学智能产业研究院(AIR)的兰艳艳老师组联合清华大学生命学院闫创业老师、张伟老师和清华大学化学系刘磊老师刚发表在《Science》上的文章,他们主要解决的是基因组级别小分子筛选的难题。

这篇文章本质上回答了这样一个问题:当我们真的想把小分子筛药做到“全基因组尺度”时,现有的方法还够不够用?
他们给出的答案并不复杂,也不炫技,但解决的是一个长期被“算力”和“规模”卡住的核心瓶颈。这也是我觉得这篇文章值得单独拿出来讲一讲的原因。
原文链接:https://doi.org/10.1126/science.ads9530
第一部分|当“小分子筛药”真的走到基因组尺度
长期以来,给蛋白找小分子配体这件事,几乎都是“逐靶点”进行的。
选一个蛋白,准备一个化合物库,跑一轮筛选,然后再换下一个靶点。
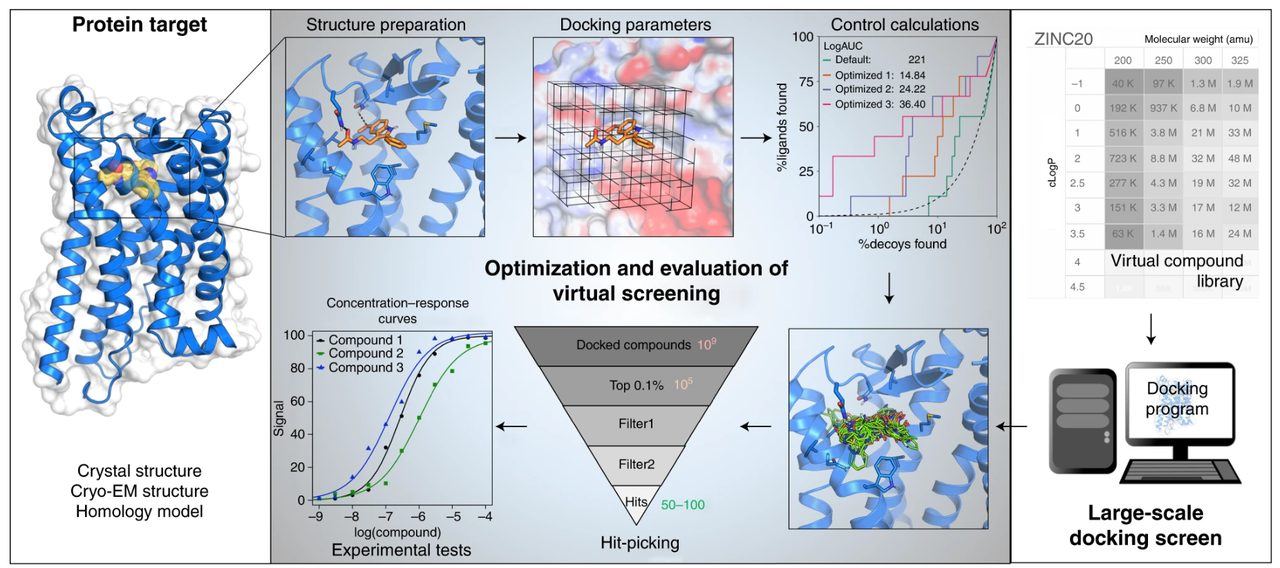
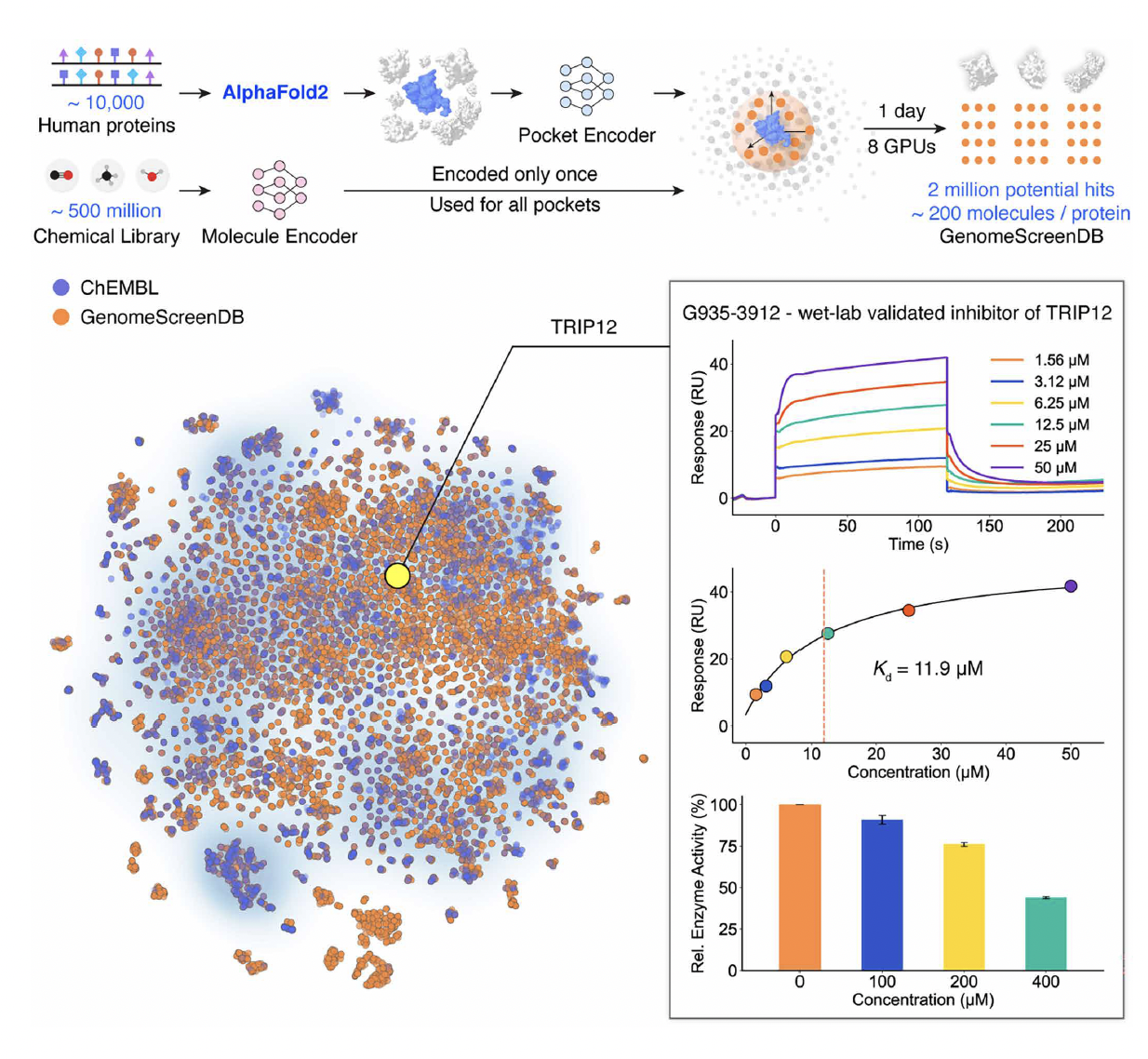
传统的小分子筛药流程
问题不在于方法,而在于尺度。
一旦把问题放到“全基因组”这个层面——成千上万的蛋白,配上百万、甚至上亿级别的小分子——传统的虚拟筛选流程就很快失去现实可行性。
所以在很长一段时间里,“基因组级别筛药”更多是一种设想:
我们知道它重要,但很少有人真的把它当成一个能跑起来的工程问题。
这篇文章做的事情,恰恰是在正面回答这个问题:
如果不再按“一个靶点一套流程”的思路来做,而是从一开始就以全基因组为目标,筛药这件事能不能被系统地做出来?
接下来他们给出的,不是一个局部优化方案,而是一整套为“规模”而设计的思路。
第二部分|用对比学习重写“筛药”这件事
为了把小分子筛选推到基因组尺度,这篇文章并没有简单地加速传统流程,而是从建模方式上换了一种思路。
他们的核心做法,是引入对比学习(contrastive learning),来学习蛋白口袋和小分子之间的匹配关系。
在这个框架里,问题不再是“这个小分子摆成什么姿势能不能 dock 进去”,
而是被重新表述为:哪些蛋白–小分子对是应该被拉近的,哪些是应该被区分开的。
模型通过大量正负样本的对比,学习一个联合表示空间:
匹配的蛋白口袋和小分子在这个空间里距离更近,不匹配的则被推得更远。
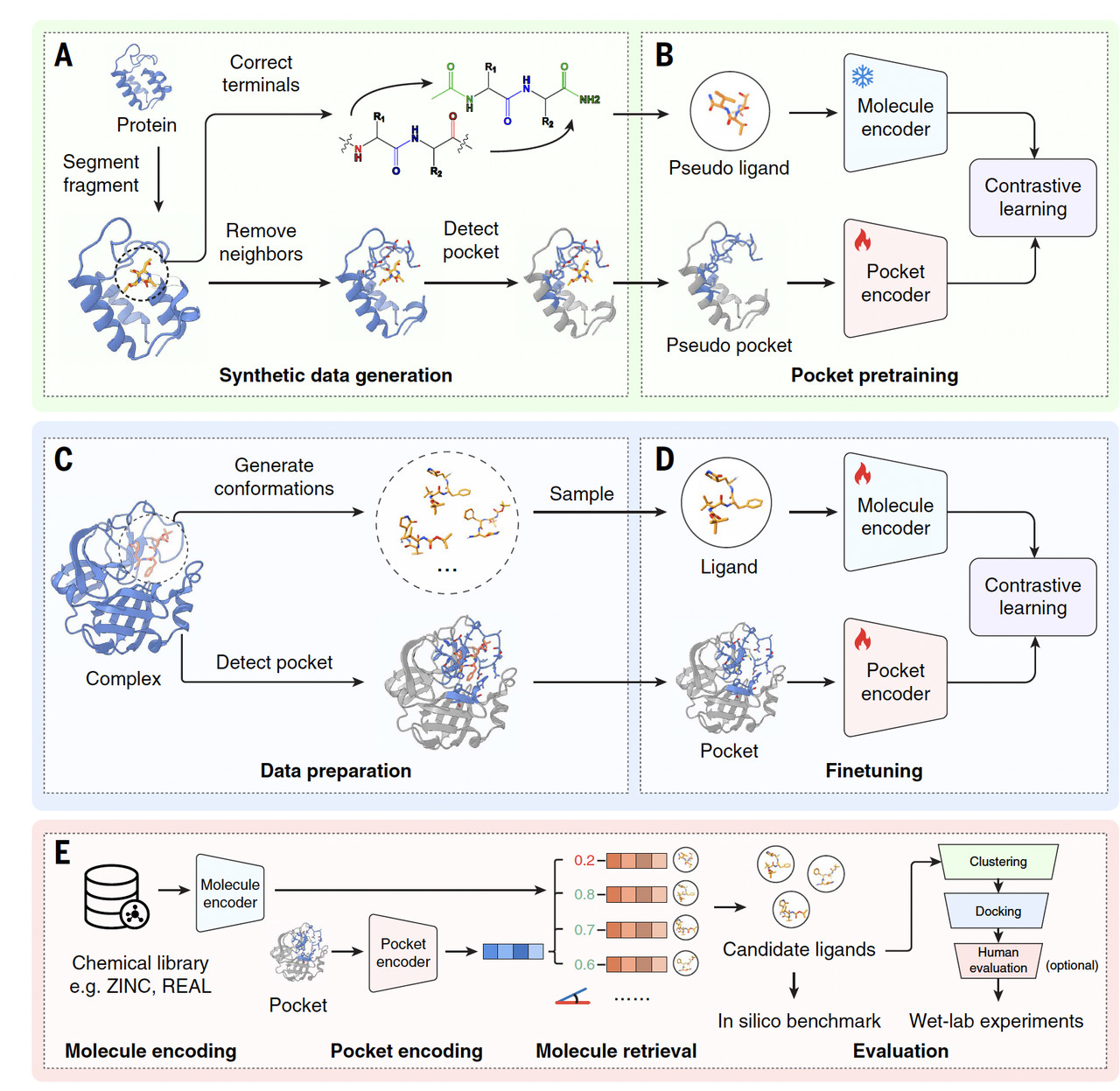
DrugCLIP模型模型架构
这一步看似只是模型训练方式的变化,但对整个问题的尺度影响非常大。
一旦这种“是否匹配”的关系被编码进表示空间,后续筛选就不再依赖复杂的构象搜索和能量计算,而可以通过高效的向量计算来完成。
也正是因为这种建模方式,作者才能把问题从“一个靶点一次筛选”,推进到“在全基因组范围内同时考虑成千上万个蛋白”。
这一步,是整篇文章能够成立的技术基础。
第三部分|不仅能跑得动,也筛得出东西
把筛选做到基因组尺度,速度只是前提。
真正关键的是:这种方法筛出来的结果,能不能经得起实验验证。
作者在这里给出了两个层次不同、但互相补充的实验案例。
第一个是 TRIP12。
这是一个并不“友好”的靶点:缺乏已解析的复合物结构,也几乎没有已知的小分子配体。作者直接使用 AlphaFold 预测结构来定义结合口袋,并在此基础上进行筛选。
最终的实验结果显示,在筛选得到的候选分子中,命中率达到 17.5%。
在“没 holo 结构、没先例”的前提下,这个结果本身就说明,这套方法筛出的并不是随机噪声,而是具备真实结合潜力的分子。
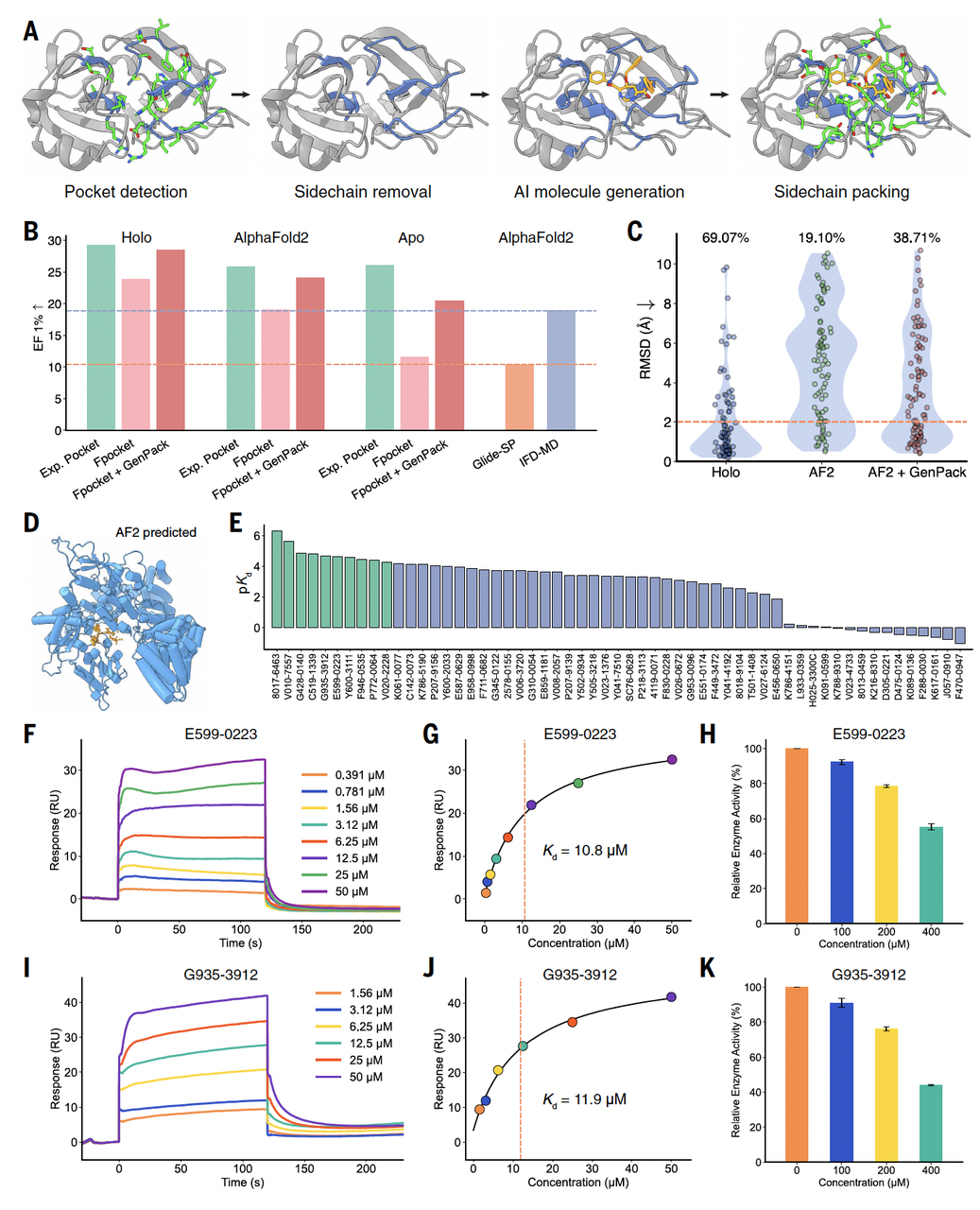
第二个案例是 NET(norepinephrine transporter)。
与 TRIP12 不同,NET 是一个已有一定研究基础的靶点,对筛选结果的区分能力要求更高,也更接近真实药物发现中的常见场景。
在 NET 上,作者同样使用模型进行大规模筛选,并对候选分子进行了实验测试。结果表明,模型给出的排序在实验上是有信息量的,活性分子能够被稳定地排在前列,而不是随机分布。
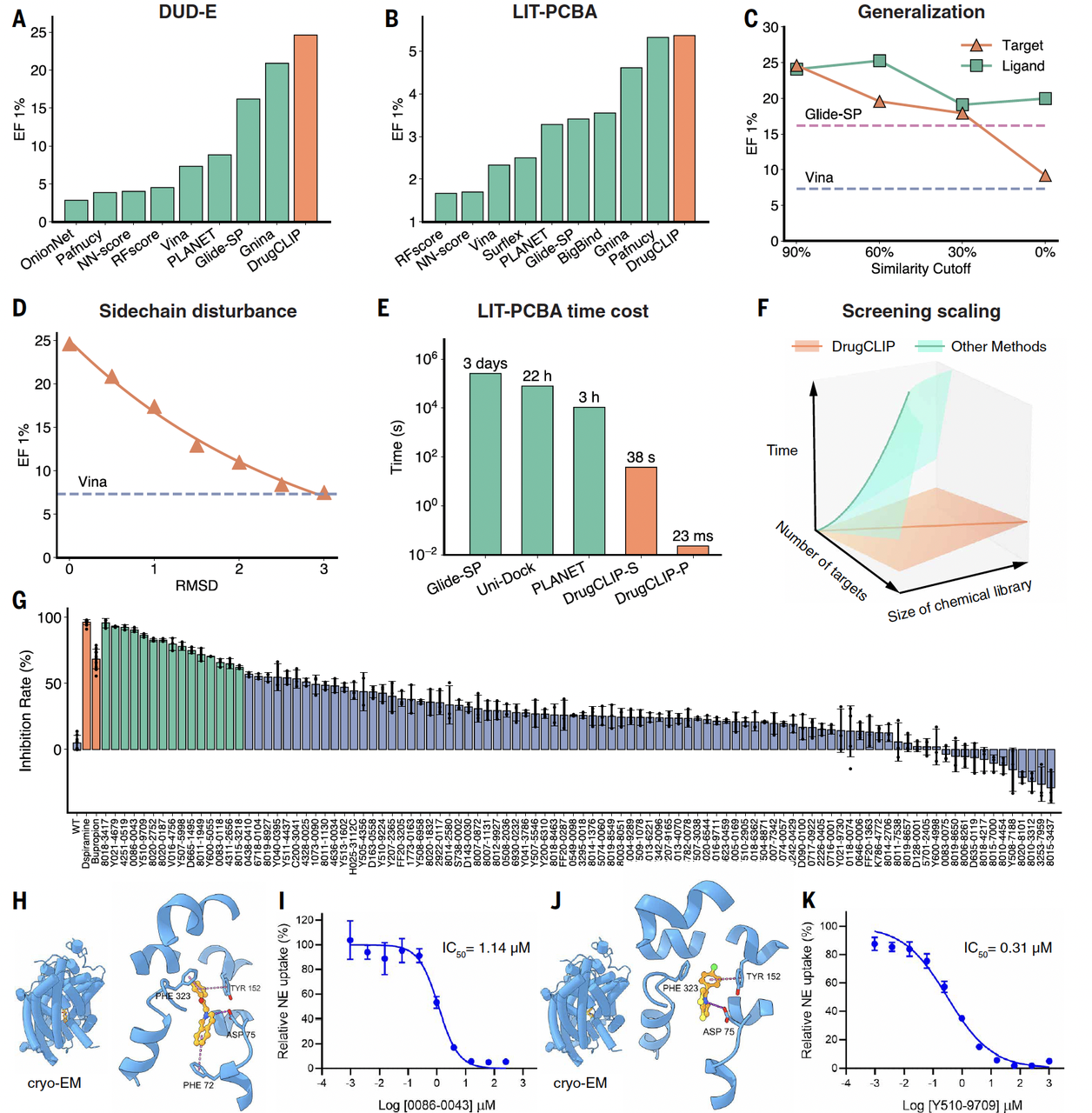
这两个例子放在一起,构成了一个很清晰的对照:
TRIP12 证明了方法在“几乎没有先验信息”的靶点上也能找到命中;
NET 则说明,在已有研究背景、需要精细区分的体系中,这套方法同样具备实用价值。
至少从实验层面来看,这并不是一套只在特殊情况下成立的筛选策略。
第四部分|把筛选结果变成所有人都能用的资源
这篇文章并没有止步于“我们证明这种方法能用”。
他们更进一步,把这件事当成一个基础设施问题来做。
作者把这套对比学习框架跑在了一个非常激进的尺度上:
覆盖大约 1 万个人类蛋白,对应 5 亿级别的小分子化合物库。
最终得到的,是一个预先计算好的筛选结果集合,并以 GenomeScreenDB 的形式对外提供。
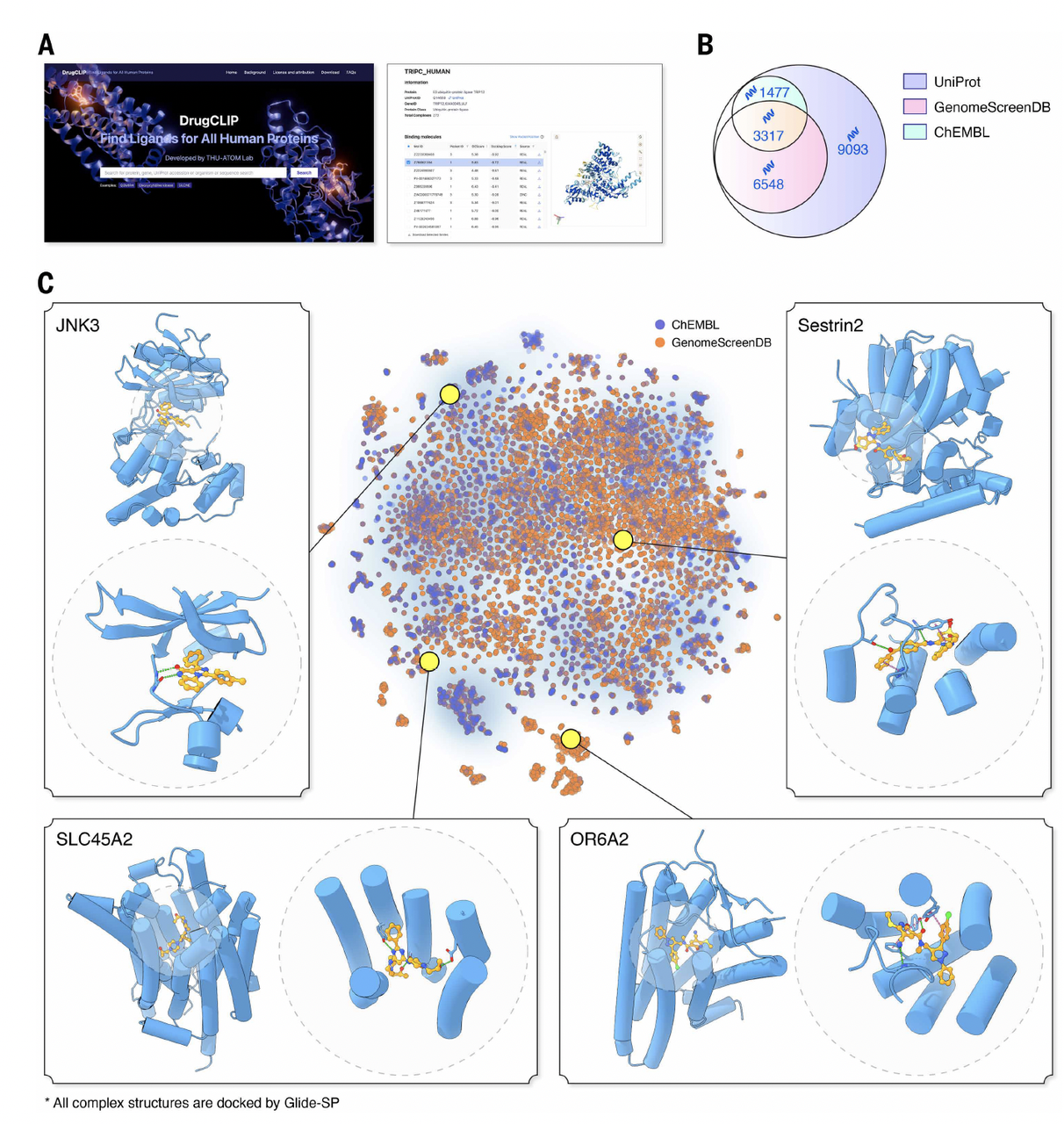
这个决定,其实很重要。
它意味着,很多时候你不需要从零开始搭筛选流程,也不需要先投入大量算力。
如果你关心某一个蛋白,或者某一类靶点,现在可以先查一查有没有已经被模型“标出来”的小分子线索。
这项工作把“基因组级别筛药”从一个方法问题,推进成了一个可直接使用的资源问题。
一点整体感受
这篇文章并不是在宣称虚拟筛选已经被解决,也不是要取代传统 docking。
它更像是在补一块长期缺失的拼图:
当你的目标是找方向、找线索、做大规模探索时,现在终于有了一条现实可行的路径。
在这个意义上,它讨论的不是某一个靶点、某一次筛选,而是
“我们能不能在全基因组范围内系统地做这件事”。
而这一次,答案至少在工程层面上,是肯定的。
来自文章作者的评价:
DrugCLIP 这一AI虚拟筛选工具相较传统方法,提升了百万倍的筛选速度,同时保持了高准确性。在日常靶点筛选中,我们已实现接近20%的粗筛阳性率,且基于AlphaFold预测的蛋白质结构,DrugCLIP同样展现了优异的阳性率。
它有效解决了庞大数据库筛选的效率问题,任何规模的筛选都能在短短半天内完成,极大提升了虚拟筛选效率,开启了药物发现的新机遇。
欢迎大家试用: https://www.drugclip.com
延伸阅读
本文属于 AI4S文献 栏目。
返回 AI4S文献 → 去公众号阅读完整版 →