RareFold:20种氨基酸不够用?让蛋白质设计进入“外挂模式”

过去,像 AlphaFold2 这样的蛋白质结构预测软件只能处理20种天然氨基酸,这就像厨师只能使用20种食材做菜,难免受限。而在自然界和实验室中,其实还存在大量“非天然氨基酸”(Noncanonical Amino Acids,简称 NCAAs),它们拥有更丰富的化学性质,比如更稳定、不易被人体分解,甚至不容易引起免疫反应,非常适合用来设计药物。
但问题是——这些“新食材”之前缺乏合适的建模工具,使用起来非常困难。
今天要介绍的这篇论文《RareFold: Structure prediction and design of proteins with noncanonical amino acids》(发表于 2025 年 5 月23日)带来了全新的解决方案:作者开发出一个可以预测并设计含非天然氨基酸蛋白结构的深度学习模型,叫做 RareFold,它不仅精准,而且效率远超前人。
接下来,我们就一起来看看 RareFold 是如何打破蛋白设计的边界的!
背景
在生命科学中,蛋白质是执行各种生物功能的“主力军”,从酶催化、信号传导,到免疫应答,几乎无所不在。而蛋白质的功能,很大程度上取决于它们的三维结构——就像工具的形状决定了用途。因此,预测蛋白质结构是理解生命机制和开发新药物的关键步骤。

过去几年,Google DeepMind 推出的 AlphaFold2 实现了蛋白质结构预测的巨大突破,精度接近实验测量,引发全球轰动。但它有一个重要局限:只能处理自然界常见的20种天然氨基酸。而在实际生物系统中,还有数百种“非天然氨基酸”(Noncanonical Amino Acids, 简称 NCAAs),它们可以带来更强的稳定性、更高的特异性,甚至更好的免疫逃逸能力,非常适合用于药物设计。
然而,由于 NCAAs 数据稀少、结构多样、现有模型无法识别,它们长期处于“有潜力但难应用”的尴尬状态。这正是本文所解决的问题 —— 如何突破传统模型的限制,把 NCAAs 纳入蛋白质结构预测与设计的工具链中。
🔬 技术亮点
RareFold 模型设计
基于 AlphaFold2 的 EvoFormer 架构扩展,支持 20 种天然氨基酸 + 29 种 NCAAs。
每种氨基酸被编码为独立“token”(包括NCAAs),使模型能学习残基特异的原子交互模式。
支持结构预测和序列-结构共同优化(即可用于蛋白设计)。
EvoBindRare 设计平台
该平台反向使用 RareFold,用于设计线性或环状肽类结合剂,并结合多种NCAAs。
成功设计出对核糖核酸酶(ribonuclease)有 μM 级亲和力的肽类(线性和环状各一),且包含非天然氨基酸。
准确性与效率
与 AlphaFold3 相比,在部分非天然氨基酸(如 MSE, SAH)上表现更优,能避免AF3中因扩散模块导致的不自然原子结构冲突。
使用 token 表示法而非原子层级操作,计算更高效:在40GB内存下可运行 AF3 无法处理的结构。
RareFold相比AF3的核心改动
1. 将氨基酸表示为离散 token
RareFold 不再依赖 AF3 的原子层级构建模块,而是将每种氨基酸(天然或非天然)都编码为一个 token,包括 20 种天然氨基酸 + 29 种 NCAAs。
这意味着每种残基都有独立的结构表示形式,允许模型学会其特定的结构和几何分布,而无需预定义原子坐标模板。
2. 调整原子映射模块
在预测结构坐标后,RareFold 使用残基特异性的原子映射规则来确定每种氨基酸的原子布局,适应非标准侧链。
AF3 的原子生成依赖模板/轨迹回归(如diffusion),但 RareFold 的方式更稳健,避免了如 MSE 残基中出现的原子冲突问题。
3. 简化模型架构,移除扩散模块
AF3 的生成式模块(如结构扩散)虽然强大,但对训练和推理资源要求极高,也容易在复杂化学结构中出错。
RareFold 去除了这些复杂部分,改为端到端结构回归 + token 解码器机制,显著降低显存需求并提高稳定性。
4. 加入支持蛋白-肽复合物建模(用于设计)
- RareFold 进一步调整了 AF3 的多链建模机制,允许预测含NCAA的蛋白-肽复合结构(特别用于 EvoBindRare 平台的设计任务)。
5. 可扩展表示结构(支持更多AA)
- AF3 中氨基酸是静态定义的,RareFold 则可以根据训练数据动态加入新的NCAA,具备更强的扩展性与迁移能力。
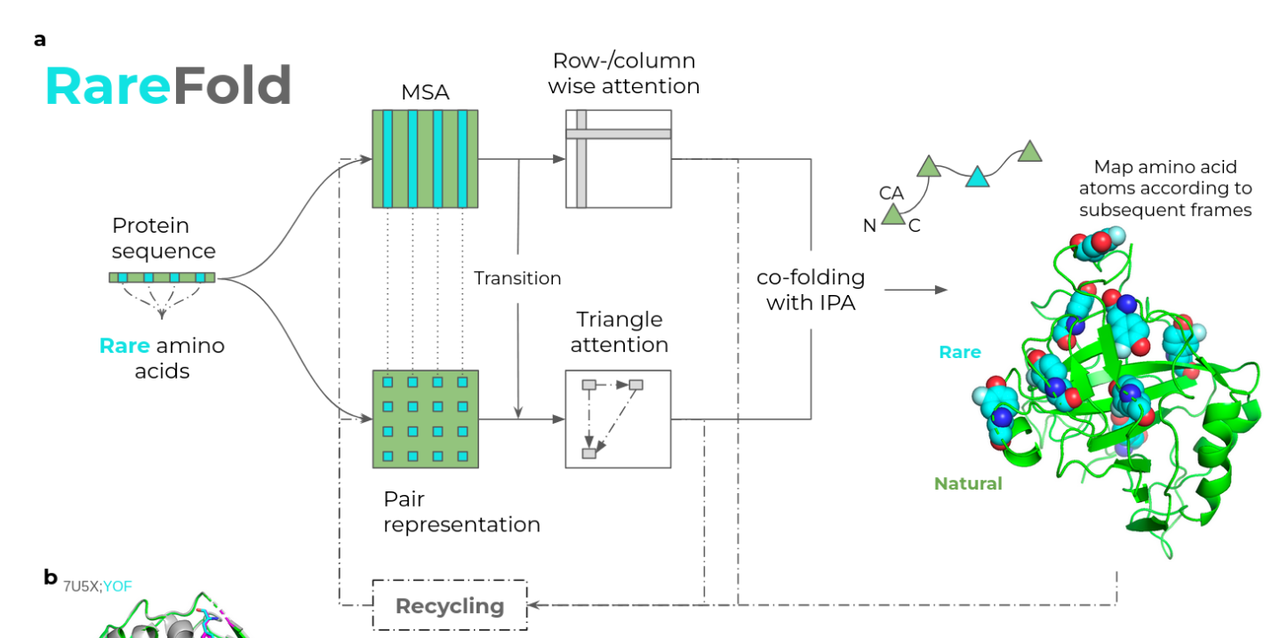
RareFold预测模型架构
RareFold整体的模型架构和AF2非常类似:
输入序列:输入的是包含“Rare”氨基酸的蛋白质序列。这些稀有氨基酸用新的 token 编码表示,打破了传统模型只支持20种天然氨基酸的限制。
共进化信息提取:
通过多序列比对(MSA)获得进化相关性。
同时构建“残基对表示”(Pair representation)用于提取结构间接关系。
结构信息建模:
依赖 AlphaFold2 中的核心注意力模块,如行/列注意力和三角注意力(Triangle Attention)来捕捉三维空间关系。
使用 IPA(Invariant Point Attention)实现蛋白质“共折叠”结构预测。
回收机制(Recycling):模型会多次迭代优化预测结果,提高准确性。
结构输出:每种氨基酸(包括非天然的)都会被分配独立的几何坐标系,从而在最终3D结构中实现精准定位和原子映射(右上图)。
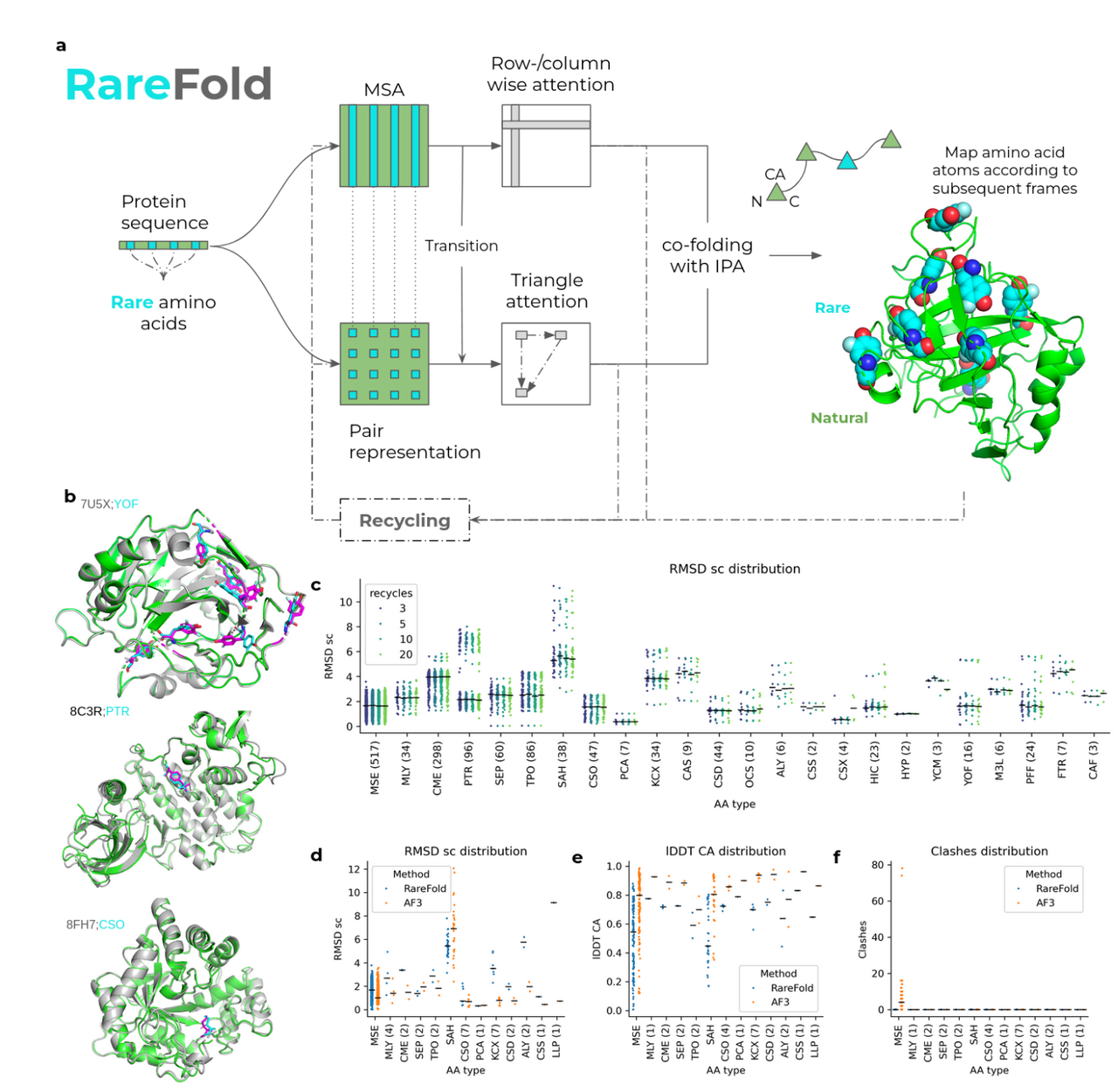
这张图详细说明了 RareFold 模型的架构、性能评估方法以及与 AlphaFold3 (AF3) 的对比,重点是对非天然氨基酸(NCAAs)结构预测的能力。
展示 RareFold 的结构预测流程:
输入蛋白质序列(包含 Rare 氨基酸)
通过 MSA(多序列比对) 和 对位表示(pair representation) 构建共演化信息
应用 行/列注意力 和 三角注意力(triangle attention) 提取结构关系
最后通过 IPA(Invariant Point Attention)对蛋白进行共折叠预测,得到3D结构
每种氨基酸(包括 NCAAs)都分配独立坐标系,确保结构精确映射
支持结构回收(Recycling) 多次迭代优化结构输出
📌 关键点:RareFold 将非天然氨基酸视为独立 token,从而学习其特定的结构模式,增强泛化能力
和alphafold2非常相似


AlphaFold3 和 RareFold谁更强?
AlphaFold3能够预测全原子结构,已经在结构预测任务中已经引入了对某些“修饰残基”的支持,例如 MSE(硒代蛋氨酸)等。因此和AlphaFold3做对比,也是RareFold 必须要做的事情。下表展示了AlphaFold3和RareFold的对比:
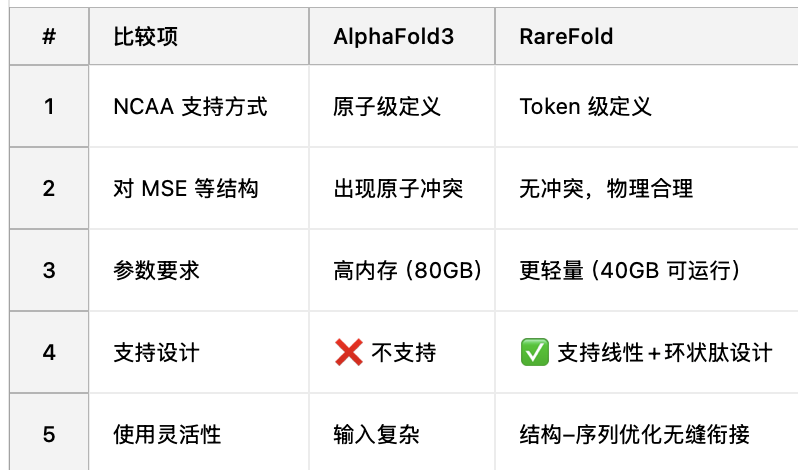
虽然 AF3 能在一定程度上处理非天然氨基酸,但它的核心设计依然是基于原子级建模 + 扩散生成模块,在处理复杂化学结构时容易出现原子冲突、建模不稳定、资源开销大等问题。而 RareFold 采用了完全不同的思路:把每个氨基酸(包括 NCAAs)看作独立 token,通过学习它们的整体结构与几何特征,实现预测过程的“抽象化”和“轻量化”。
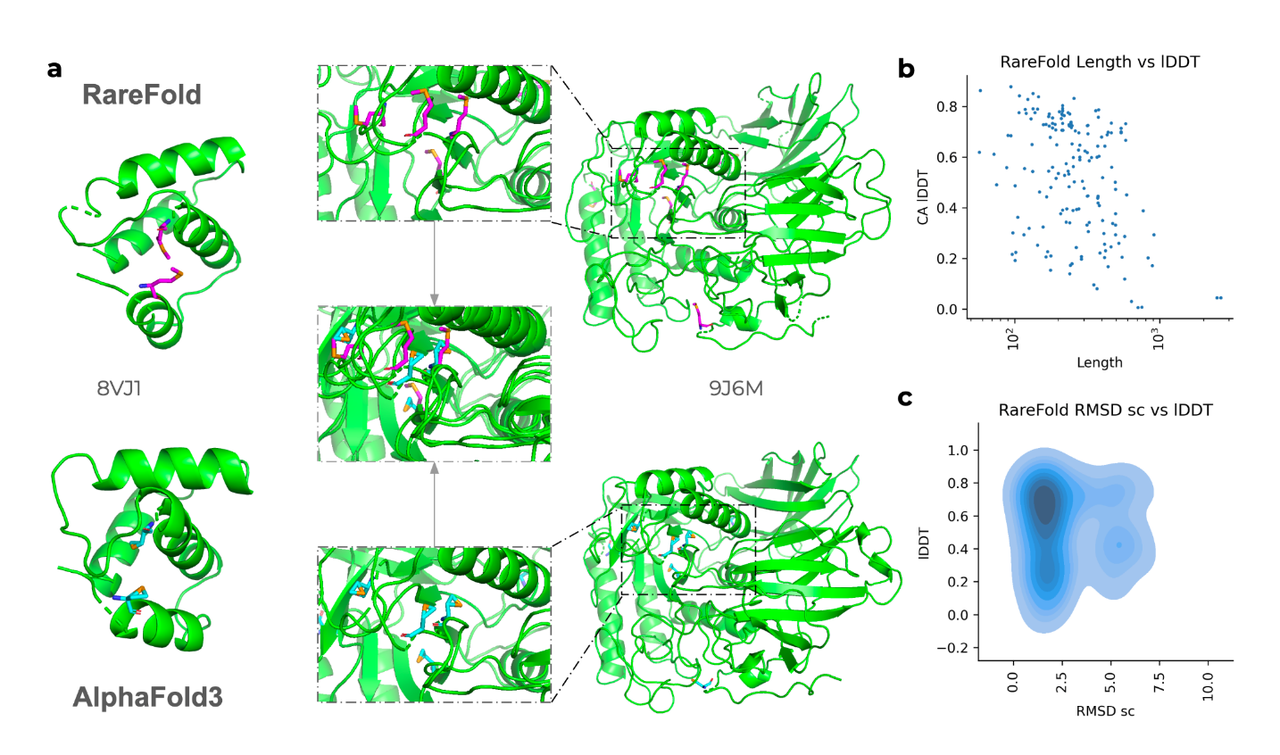
这张图主要展示了 RareFold 相比 AlphaFold3 在预测非天然氨基酸(NCAA)结构时的表现,具体包括结构准确性、预测置信度和蛋白长度对结构预测精度的影响。
图 a:RareFold vs AlphaFold3 对 MSE 残基的结构预测
展示的是蛋白 8VJ1 和 9J6M 中的 MSE(硒代蛋氨酸) 的结构预测比较。
绿色为主链结构,粉色/青色棒状为 MSE 残基的侧链原子。
上排是 RareFold 的预测,MSE 结构合理且无冲突;
下排是 AlphaFold3 的预测,可以看到存在原子间重叠(冲突),尤其是 MSE 中的硒原子与相邻原子过于接近,导致物理结构不合理。
📌 结论:RareFold 相较于 AlphaFold3 能更好地处理 MSE 这类NCAA,预测出的结构更物理合理,避免了常见的原子间冲突问题。
图 b:RareFold 的预测准确度(Cα lDDT) vs 蛋白长度
X轴为蛋白序列长度(对数刻度),Y轴为预测结构的 Cα lDDT 分数(预测结构与真实结构的相似度,越高越好)。
数据显示:随着蛋白长度增加,预测准确度略有下降,但并无明显断崖式下降。
📌 结论:RareFold 在预测较长序列时会面临一定挑战,但整体仍维持较高准确度,说明模型具有较强的泛化能力。
图 c:RareFold 的 NCAA 侧链预测误差(RMSD) vs IDDT
横轴是 NCAA 的侧链 RMSD(预测与真实之间的误差,越小越好),纵轴是 IDDT(全局结构准确度)。
图像是一个 密度图,深蓝区域表示数据点密度高。
可以看到,在 结构整体准确(高 IDDT)时,侧链误差也通常更小。
📌 结论:整体结构预测质量(IDDT)与局部侧链精度(RMSD)之间存在正相关关系;即当结构整体预测得好时,非天然氨基酸的预测也更准确。
✅ RareFold 能预测得准吗?看准度也要看“自信心”(氨基酸准确性和置信度)
AI 模型不仅要预测准,还要知道“自己预测得准不准”——也就是说,它的置信度得分(plDDT)要能真实反映结构质量。RareFold 在这方面表现如何?这张图给出了非常直观的答案:
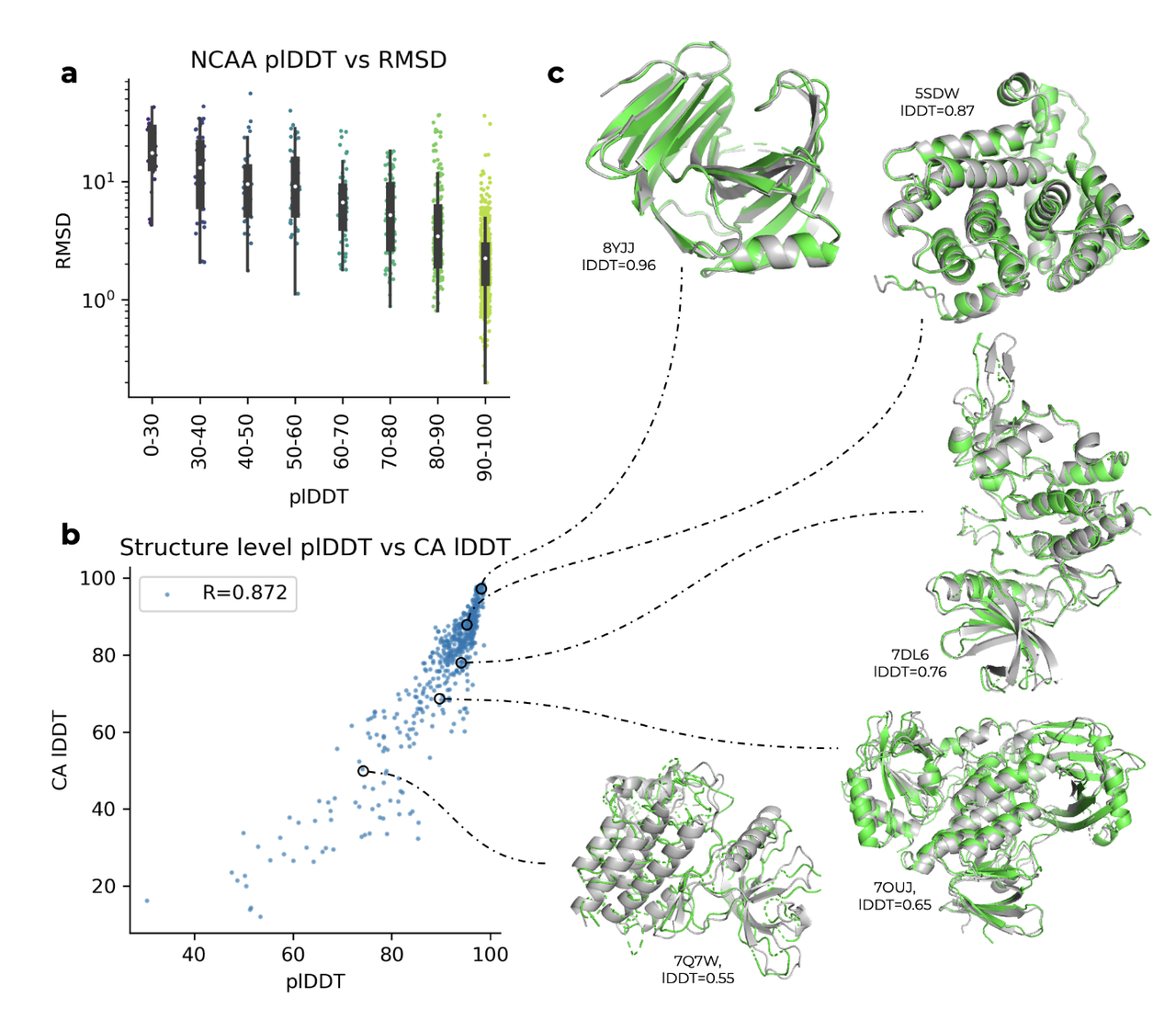
核心结论:
图 a 总结:plDDT 越高,非天然氨基酸的结构预测误差(RMSD)越低,说明模型能判断预测是否可靠。
图 b 总结:整个蛋白结构的平均置信度(plDDT)与真实结构相似度(IDDT)高度相关,R=0.872,反映整体预测可信度高。
图 c 总结:高 IDDT 的预测结构与真实结构几乎重合,低 IDDT 则偏差明显,视觉上验证了模型置信度与准确度的一致性。
图 a: plDDT vs RMSD(单个氨基酸层面)
横轴: plDDT(模型对每个残基的预测置信度,0–100)
纵轴: RMSD(预测与真实结构的误差,log尺度)
每个点表示一个 NCAA 残基
🔍 观察结果:
plDDT 越高,RMSD 趋势越低:说明 plDDT 可以用来判断预测是否准确。
大多数预测位于 90–100 的高置信区间,共有 896 个点,RMSD 也较低,表示预测可信。
📌 结论: RareFold 能够通过 plDDT 成功识别哪些残基预测得准、哪些不准。
图 b: plDDT vs Cα IDDT(全蛋白层面)
横轴: 平均 plDDT 分数(整个结构)
纵轴: Cα IDDT(预测结构与真实结构在主链 α-碳原子上的相似性)
每个点代表一个完整蛋白结构
🔍 观察结果:
两者呈高度正相关,相关系数 R = 0.872,即置信度高的结构预测也更精确。
大多数结构分布在高区间(右上角),说明整体预测质量高。
📌 结论: RareFold 的置信度评分不仅在单个氨基酸层面有效,也在整链结构层面准确反映预测质量。
图 c: 可视化展示不同 IDDT 水平下的结构预测
展示了几个具有代表性的蛋白质预测与原始结构的对比(绿色为预测结构,灰色为真实结构):
8YJJ (IDDT=0.96):几乎完美预测
5SDW (0.87)、7DL6 (0.76):较好预测
7QUW (0.55)、7OUJ (0.65):预测效果下降,误差明显
📌 结论: 高 IDDT 的预测结果与真实结构非常接近,进一步验证 RareFold 的置信度评分与预测精度高度一致。
🔍 RareFold 对非天然氨基酸的预测到底准不准?
既然 RareFold 主打“能看懂非天然氨基酸”,那它在实际预测中表现如何?作者通过大规模测试,把 RareFold 在不同氨基酸上的预测结果做了统计,从侧链误差(RMSD)和整体结构准确度(IDDT)两个维度,分别和天然氨基酸进行了对比。
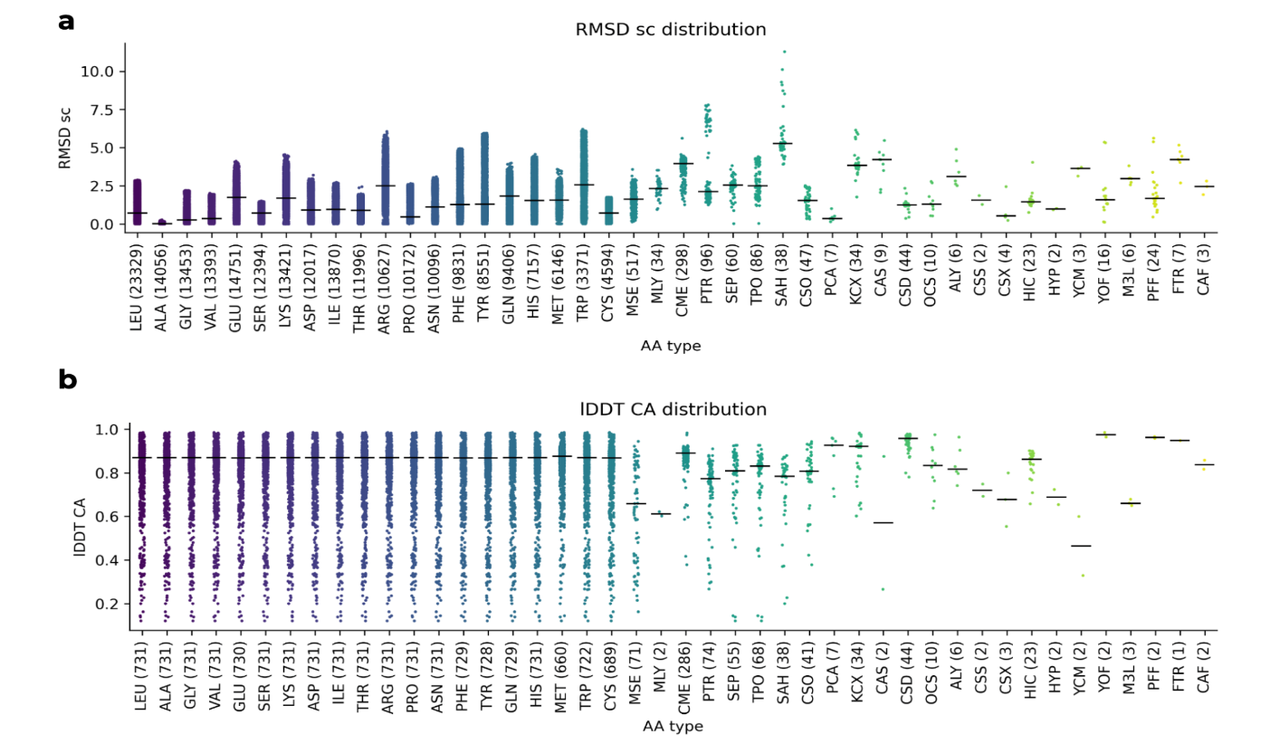
结果很令人安心:
在预测精度上,像 MSE、SEP、PTR 等常见的非天然氨基酸,其 RMSD 表现和 LEU、ALA 等天然氨基酸几乎处于同一水平线,误差非常小,说明模型对这些结构“非常熟”。
即便从整条蛋白质的角度看,RareFold 在这些 NCAAs 上也能预测出高 IDDT 的主链结构,和天然氨基酸一样靠谱。
虽然一些极其稀有、样本极少的 NCAAs(比如 CAF、PFF 等)会存在波动,但这属于“训练数据稀缺”的客观现象,并不影响整体可靠性。
📌 一句话总结:RareFold 预测大多数非天然氨基酸的精度,已经足以和天然氨基酸一较高下,不再是“AI 看不懂的稀有语言”。这为后续结构设计和药物开发打下了坚实基础。
这张图(图4)展示了 RareFold 在结构预测中对天然氨基酸(proteinogenic AAs)和非天然氨基酸(NCAAs)的表现比较,重点分析的是结构预测误差(RMSD)和准确度(lDDT)在不同氨基酸类型上的分布情况。
📊 图 a:不同氨基酸侧链 RMSD(结构误差)分布
横轴:氨基酸类型(从左到右依次为天然 → 非天然)
括号内数字:该氨基酸在验证集中的出现次数
纵轴:侧链 RMSD(sc RMSD),越低表示预测越精确
🔍 解读:
天然氨基酸(如 LEU, ARG, THR)通常 RMSD 较低,但也有一定波动。
非天然氨基酸(如 CME, SEP, PCA)整体误差稍大,但差异可接受。
某些非天然氨基酸样本数很少(如 CAF 仅3个),误差波动较大。
📌 结论:
RareFold 对非天然氨基酸的预测误差在可接受范围,如 CME 与 ARG 的 RMSD 分布范围类似,说明该模型能较好地泛化到NCAAs。
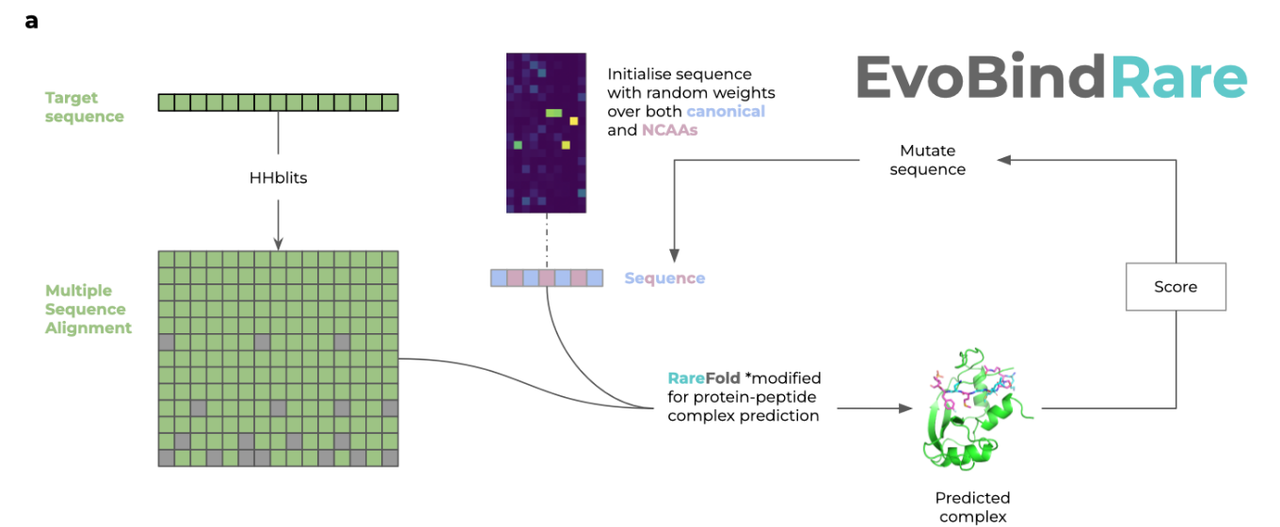
🧬 用 RareFold 来反向设计蛋白:EvoBindRare 是怎么做到的?
结构预测只是第一步,更激动人心的是——RareFold 不只“看懂”了非天然氨基酸(NCAAs),它还能反过来参与设计!这就是本文提出的 EvoBindRare 系统。
其实 EvoBindRare 本质上是之前工具 EvoBind2 的一次“插件升级”——原本使用 AlphaFold 作为预测模块,现在换成了 RareFold,因此就具备了处理 NCAAs 的能力,实现了对线性和环状肽结合剂的设计支持。
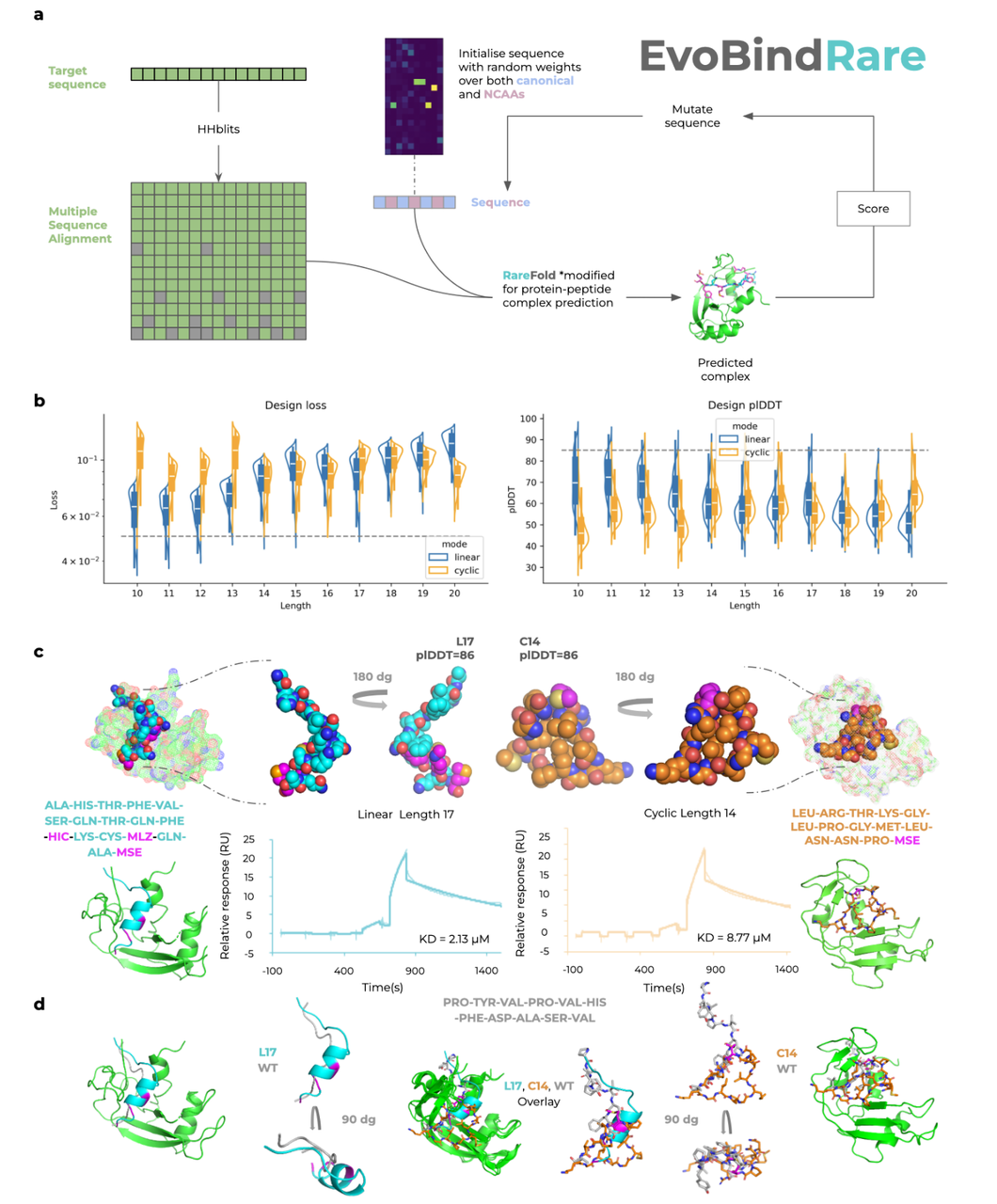
这张图(图5)展示了 RareFold 模型在蛋白质设计中的应用,特别是其子系统 EvoBindRare(EBR) 用于设计含非天然氨基酸(NCAAs)的肽类结合剂(peptide binders)的完整流程、性能评估和实验验证。
🧬 图 a:EvoBindRare 的设计流程(流程图)
展示了 EBR 的逆向设计架构:
输入目标蛋白质序列 → 使用 HHblits 生成 MSA(多序列比对)
初始化肽序列(包含天然和非天然氨基酸) → 随机起始序列
结构预测模型 RareFold(改为预测蛋白-肽复合物)对该序列进行结构预测
计算结构评分(loss)→ 用于指导序列的突变优化
$$\text{Loss}1 = \text{peptide } plDDT^{-1} \cdot \left( \frac{1}{n} \sum{j=1}^{n} d_j \right) + % \text{clashes} $$
- 不断迭代突变并打分,优化出具有高亲和力的肽结合剂
📌 亮点:该流程允许引入 NCAAs,拓展设计空间,最终实现高效肽类药物设计。
📊 图 b:设计性能评估(设计损失与置信度)
左图:设计损失(越低越好)随肽长度变化(X轴:10–20肽长)
右图:预测置信度(plDDT)随肽长度变化(Y轴越高越可靠)
颜色区分:
蓝色为线性肽
橙色为环状肽(更难设计)
🔍 结论:
大多数肽长都能设计出低loss且高plDDT的结构;
环状肽的plDDT略低,说明结构约束更复杂;
多数设计超越了置信度阈值(plDDT > 85)和损失阈值(loss < 0.05)。
📌 亮点:EvoBindRare 可以系统性生成可靠的多种肽结构设计。
🧪 图 c:成功设计并验证的两个结合肽(线性 & 环状)
展示了两个结合肽的设计、预测结构和实验结合验证:
左侧为线性肽(17肽长):包含多个 NCAAs(如 MSE, MLZ, HIC),plDDT=86,Kd = 2.13 μM
右侧为环状肽(14肽长):也包含 NCAAs,plDDT=86,Kd = 8.77 μM
底部为 SPR 实验曲线,验证肽与目标蛋白结合的亲和力(μM 级)
📌 亮点:设计的肽不仅在结构上可靠,还在实验中表现出有效结合活性,证明 RareFold+EBR 的功能性。
🧬 图 d:预测结构与已知结构比对(WT vs 设计肽)
左:线性肽 L17 与已知天然肽(WT)叠加比较 → 二者结构相似但结合方式不同;
右:环状肽 C14 与 WT 比较 → 呈现出不同但合理的结合构象;
📌 亮点:EBR 不依赖模板,能设计出与天然结合肽结构不同但功能等效的新型肽。
✅ 总结
这张图展示了 RareFold 的设计模块 EvoBindRare:
能设计含NCAAs的高亲和力肽;
成功生成线性和环状结合肽;
并通过实验验证了其预测的功能性。
Data Availability: https://zenodo.org/uploads/14892196.
Code Availability:https://github.com/patrickbryant1/RareFold
方法细节:
数据:
数据来源:
所有蛋白质结构均来自 PDB 数据库,提取时间为 2024年12月10日
仅选择通过 X射线衍射或电子显微镜(EM)测定,分辨率 ≤ 5 Å 的单体蛋白结构
总共选取了 75,232 条结构
筛选标准:
每个结构文件只提取 第一个蛋白链
保留序列中非规则氨基酸(包含NCAAs)比例小于80%,序列长度大于50
满足以上条件的样本数为 74,882 条(占总数 99.5%)
🧪 序列聚类处理
为了降低冗余度,使用 MMseqs2 工具 对序列按 20% 相似性 进行聚类
最终得到 9,031 个代表性簇
非天然氨基酸的描述
RareFold 需要对这些 NCAAs 进行结构预测,但 AlphaFold2 仅支持标准的 20 种天然氨基酸,因此作者扩展了结构表示框架,引入了新的残基原子坐标模板。


多序列比对
NCAAs 无法用于标准 MSA 编码
多数非天然氨基酸没有标准的一字母代码,因此无法直接参与多序列比对(MSA)。
只有极少数 NCAAs 被编码为特殊字符:
SEC (硒代半胱氨酸) → ‘U’
PYL (吡咯赖氨酸) → ‘O’
GLX (模糊的谷氨酰/天冬酰胺) → ‘X’(等价于未知/UNK)
🔍 但问题在于:
‘X’ 会被解读为“未知残基”(UNK),信息价值几乎为零
‘U’ 和 ‘O’ 出现在实际蛋白数据中非常少(不在前50常见残基中)
✅ 解决方法:将所有非标准氨基酸替换为 ‘X’
为进行 MSA 构建:
所有非标准残基(NCAAs)都被统一替换为 ‘X’
这样虽然牺牲了特异性,但保留了比对流程的兼容性
MSA 计算的瓶颈与加速策略
RareFold 仅使用 uniclust30_2018_08 数据库
工具为 HHblits(HH-suite v3.1.0)
使用的命令行为:
训练与验证集
为比较 RareFold 与 AlphaFold3 在预测非天然氨基酸结构方面的性能,作者构建了一个包含修改残基(NCAA)的训练和评估数据集。
划分标准:
将 PDB 中的数据以 2021-09-30 为时间界限,这一天也是 AF3 的训练数据截止时间。
所有早于此日期的结构用于训练;
晚于此日期且未出现在AF3训练集中的结构用于测试;
其余用于验证。
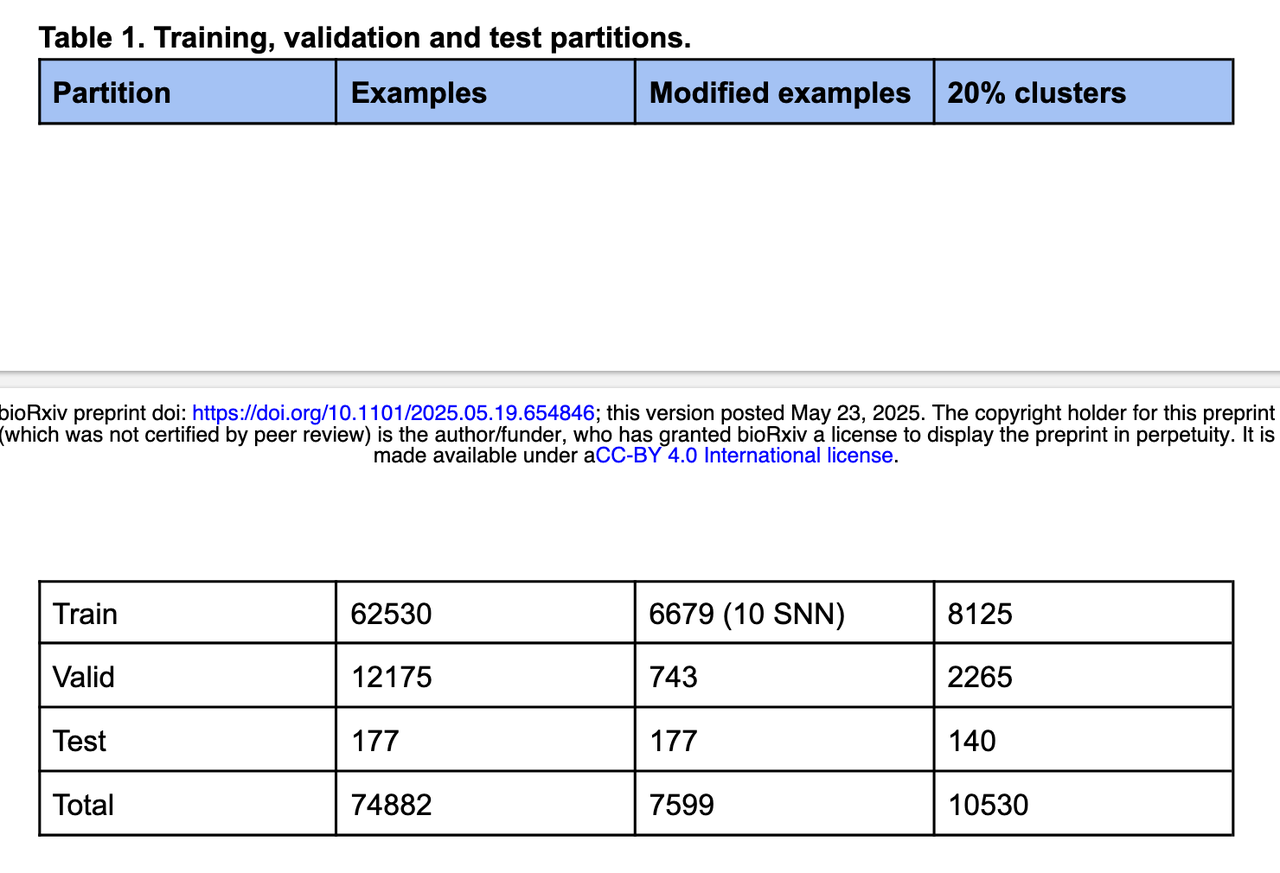
训练集:
总计 62,530 个结构
其中 6,679 个包含非天然氨基酸(包含10个 SNN)
覆盖 8,125 个 20% 相似性聚类
测试集:
从时间分割后新出现的结构中选出 177 个包含非天然氨基酸的结构
这些结构不在 AF3 训练集中,适合作为独立对比基准
验证集:
包含 12,175 个结构
其中 743 个含有 NCAA
用于训练中模型性能监控
训练
训练数据构成(非天然氨基酸优先):
每个训练 batch 的一半样本必须包含非天然氨基酸。
这些样本根据其在数据集中出现的“稀有程度”采样,确保模型能学习到罕见NCAAs的结构特征。
另一半样本来自代表性蛋白序列(基于 20% 序列相似度簇),以增强模型泛化能力。
训练配置:
batch size = 24
在 8 张 A100 GPU(每张80GB显存)上进行训练
总训练步数:40,000步(前20,000步标准训练,后5,000步加上额外loss进行精调)
损失函数定义:
总损失由以下项加权组成:
$$\text{Loss} = 0.5 \cdot \text{FAPE} + 0.5 \cdot \text{AUX} + 0.3 \cdot \text{Distance} + 0.2 \cdot \text{MSA} + 0.01 \cdot \text{Confidence}$$
其中:
FAPE:结构原子对齐误差(核心结构损失)
AUX:由 FAPE 和角度误差组成的辅助损失
Distance:残基间距离损失
MSA:掩码多序列比对预测误差
Confidence:预测 lDDT 与真实值之间的差异
这些损失项完全复用自 AlphaFold2 的定义。
微调
精调(Fine-tuning)阶段的目的是进一步优化模型输出结构的物理合理性,减少原子冲突和结构违规,使得模型更适合反复调用的蛋白质设计任务。
❗ 为什么要精调?
模型初步训练后,可能存在结构中的冲突或物理违规,例如:
残基之间的键长异常
主链 Cα–Cα 距离过远或过近
残基间原子发生碰撞(clashes)
这些问题在设计场景中必须避免,否则每次调用都要额外执行能量松弛(relaxation),计算开销巨大。
✅ RareFold 的精调策略
使用与 AlphaFold2 相同的损失函数与权重,专注于以下违规类型:
残基级违规(violations per residue)
残基之间键长违规(between-residue bond violations)
Cα–Cα 距离违规(extreme Cα–Cα distance violations)
残基间原子冲突(between-residue clashes)
不包括残基内部(intra-residue)违规的精调(因为这类错误影响相对较小或更难优化)
精调共进行 5,000 步,加上之前的 20,000 步总计 25,000 步训练,训练样本数为 600,000
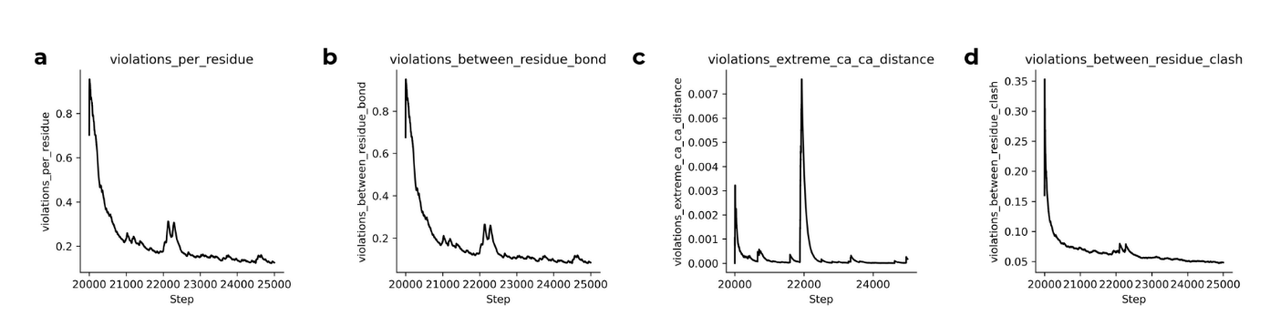
结构松弛
🌿 什么是 Relaxation?
Relaxation 是一种 分子动力学(MD)模拟 技术,用于:
缓解残基间的原子冲突(inter-residue clashes)
改善局部几何结构,使预测结果更加物理合理
🛠 技术细节
使用工具:OpenMM(开源分子模拟平台)
力场参数:CHARMM36 力场
积分方法:Langevin middle integrator(适用于温控条件下的分子模拟)
🔄 非天然氨基酸如何处理?
由于 OpenMM 不原生支持非天然氨基酸(NCAAs):
模拟时,将NCAAs暂时替换为最相近的天然氨基酸(canonical analogues)
结构优化完成后,再将这些残基换回原始的 NCAAs
⚠️ 影响评估
结构形状不会显著变化(即 relaxation 不会破坏原始预测结构)
但 部分评估指标(如 RMSD、IDDT)会轻微下降
- 因为模拟优化的微小移动可能让预测值略偏离原始 ground truth
验证方法和结果
✅ 验证流程
每隔 10,000 步在训练过程中进行一次模型验证:
- 评估点包括训练步数 20,000、30,000、40,000 和 fine-tuning 后
验证使用的是 743 个包含修饰氨基酸的样本(来源于 Table 1)
📌 评分成功率:
成功打分的结构有 731 个(98.3%)
有 12 个样本因结构不一致被排除
📌 松弛(relaxation)后表现:
在 step 20,000 应用的结构松弛步骤对整体准确性影响不大
但由于 OpenMM 中缺乏部分非天然氨基酸的原子力场定义,只有 679 个结构(93%) 成功完成松弛
打分
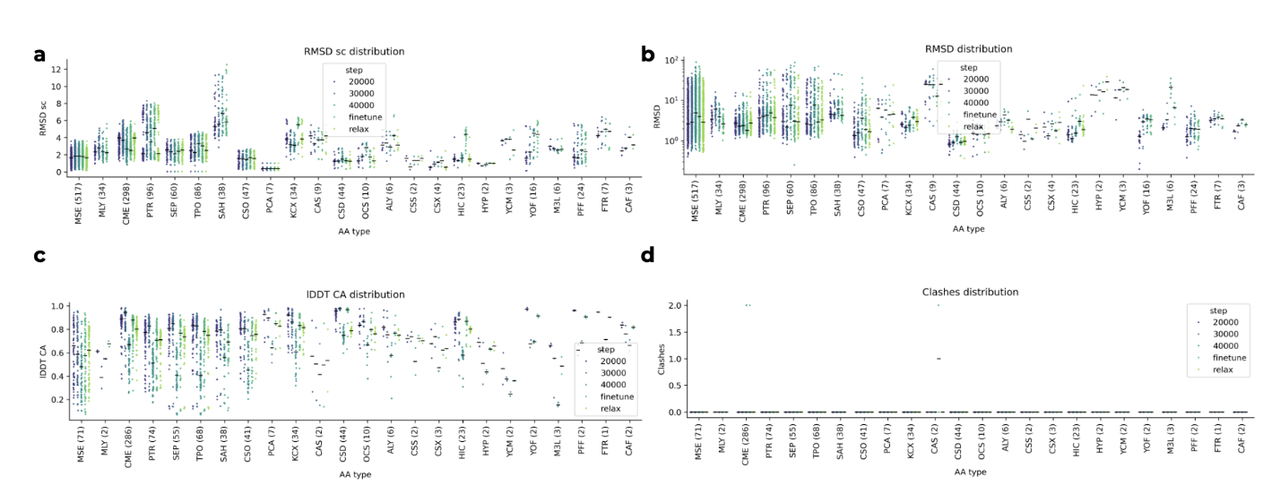
🔹 结构对齐与 RMSD 计算
首先将预测结构与真实结构在 Cα 原子(主链核心)上对齐
然后对每一个被修改的氨基酸(mod AA,即非天然氨基酸):
整体 RMSD:考虑该残基的所有原子(包括主链和侧链)
侧链 RMSD(RMSD sc):主链原子(N, Cα, C)对齐后,仅计算侧链原子的 RMSD
🔹 全局结构评分
- 使用 Cα IDDT 分数(lDDT CA) 衡量整个结构的准确性(分数越高表示与真实结构更接近)
RareFold 的反向设计(inverse design)流程
创建了一个名为 EvoBindRare 的平台,通过 RareFold 的结构预测能力,设计能与特定蛋白结合的肽(binder)。
使用的氨基酸集合:
包含20种天然氨基酸
加上12种实验上可合成的非天然氨基酸(NCAAs)

初始化与优化过程:
每条肽从随机序列初始化;
使用下面这个 loss 函数对肽结构进行迭代优化(1000次迭代):
$$\text{Loss}1 = \text{peptide } plDDT^{-1} \cdot \left( \frac{1}{n} \sum{j=1}^{n} d_j \right) + % \text{clashes} $$
每次设计任务运行 1000次迭代,使用24种不同初始化序列
支持10~20残基长度,设计线性或环状肽
每个设计使用 3次结构回收(recycle)
使用改写后的 RareFold 模型,可以并行设计24种肽,只需一张 NVIDIA A100 GPU(40GB 显存)
使用简介
RareFold 能够预测包含稀有非天然氨基酸的单链蛋白质结构,并通过 EvoBindRare 框架实现新型肽类结合剂的设计。
RareFold 支持共 49 种氨基酸类型,包括:
20 种常见天然氨基酸;
29 种稀有非天然氨基酸,包括:
MSE、TPO、MLY、CME、PTR、SEP、SAH、CSO、PCA、KCX、CAS、CSD、MLZ、OCS、ALY、CSS、CSX、HIC、HYP、YCM、YOF、M3L、PFF、CGU、FTR、LLP、CAF、CMH、MHO
EvoBindRare 可直接从目标蛋白序列设计线性或环状结合肽,无需预先知道结合位点(但可选提供)。该框架通过引入非天然氨基酸,显著拓展了可用于设计的化学多样性,支持快速且灵活的肽类分子设计。
Prediction:
Input:
fasta
num_recycles
outdir
Output:
- pdb
Design:
Input:
num_recycle
binder_length
num_iterations
resample_every_n
batch_size
rare_AA
cyclic_offset
save_best_only
outdir
延伸阅读
本文属于 AI4S文献 栏目。
返回 AI4S文献 → 去公众号阅读完整版 →