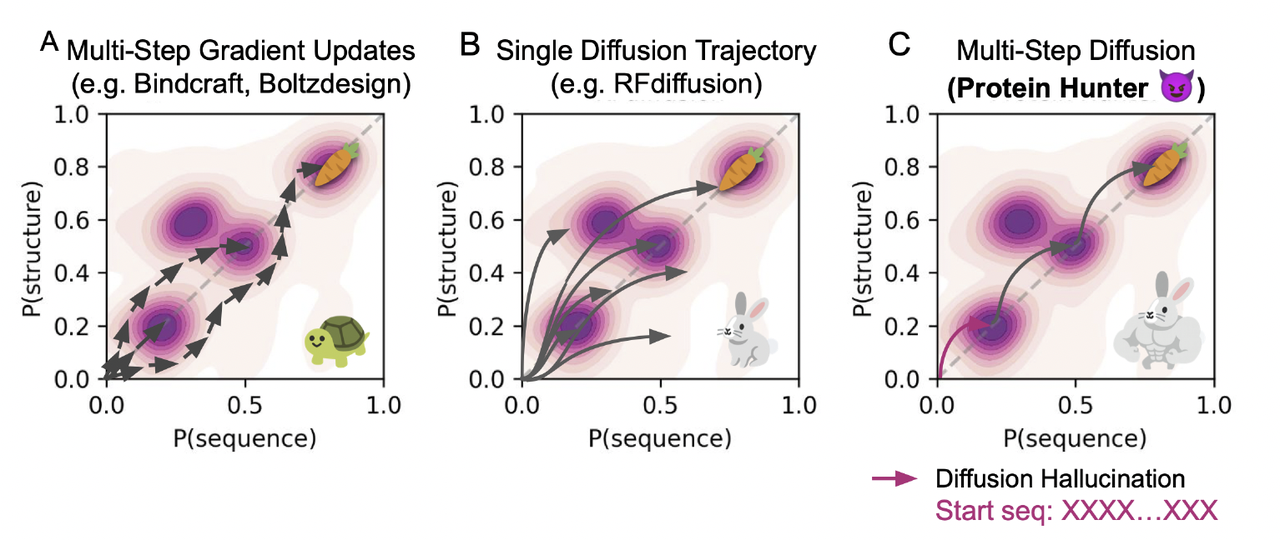
Protein Hunter 深度解读:利用结构预测模型“幻觉”构建通用蛋白设计引擎
有时候,蛋白质预测模型的“幻觉”已经足够“天马行空”,我们为什么不直接利用模型足够强大的能力,让结构预测先先天马行空地“做梦”,再把“做的梦”打磨成真实的蛋白。今天要讲的Protein Hunter 就这样,用幻觉打开了蛋白设计的新入口。
一:从“幻觉”现象出发——将结构预测模型转化为结构生成器
近年来的结构预测模型(如 AlphaFold3、Boltz)在处理“信息极度不足”的输入时,会呈现出一种稳定的“hallucination”行为:即使序列缺乏明确的进化线索,它们仍能收敛到一套自洽且具有天然蛋白典型特征的三维构象。
传统观点将此视为模型的“非物理泛化”,但今天要讲的这篇论文《 Protein Hunter: exploiting structure hallucination within diffusion for protein design》的出现,这一特性被重新定位——不是缺陷,而是一种可利用的结构生成能力。

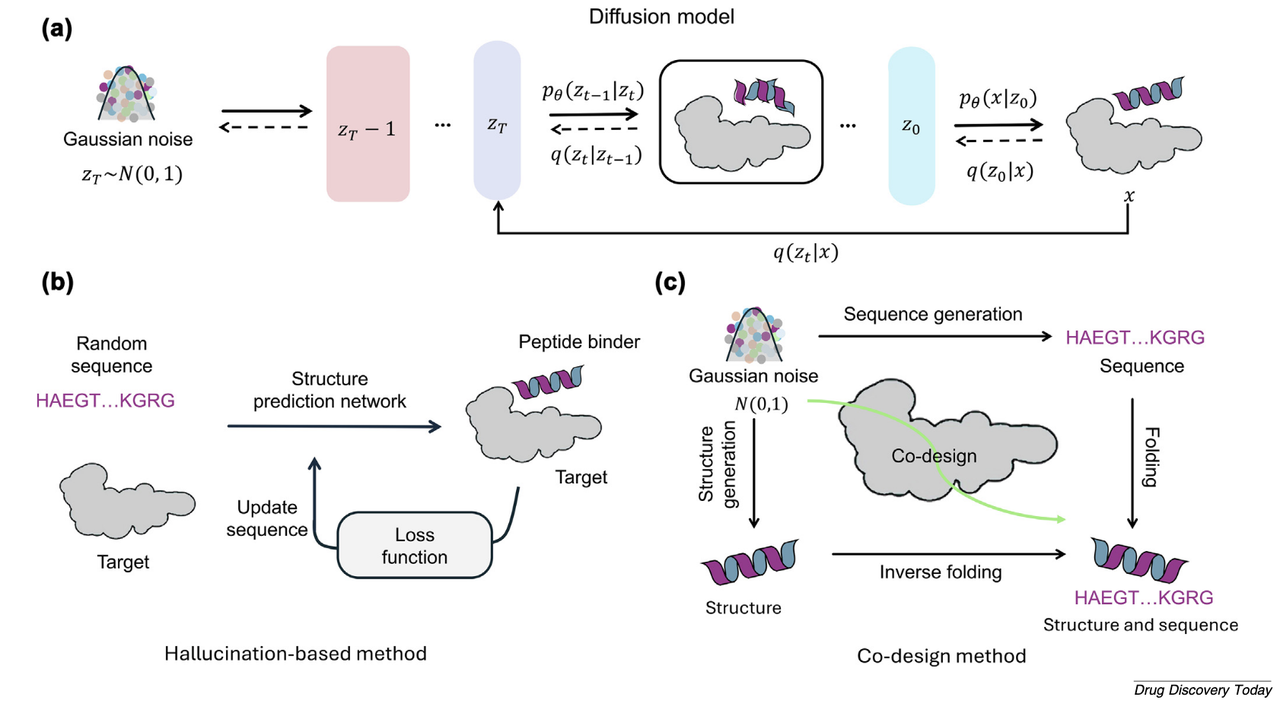
结构预测模型的扩散过程本质上已经包含“从粗到细的构象重建”
以 Boltz 为代表的 AF3 风格模型内部包含多步迭代:模型先给出一个较粗糙的初始构象,然后在多轮推理中不断修正局部几何、优化骨架一致性,并逐步形成完整的折叠模式。
如果从这一机制角度重新审视模型的推理路径,会发现它与生成模型的扩散轨迹并不矛盾:
结构预测本身已经隐含了一条“从随机构象到合理折叠”的动态路径。
论文中Figure 1正是基于这种理解:作者不再将结构预测视为一个单点函数,而是将其看成一个可以“引导构象收敛”的隐式生成器。
关键策略:从一条“全 X 序列”出发,让模型自由地构建骨架
这篇工作的核心创新非常直接但颇具突破性:
研究者给模型输入一条完全由 X token 组成的序列——在语义上等价于“每个位置的氨基酸未知”。
在常规直觉里,这应该导致模型输出一个不稳定或无意义的结构;然而由于上述的 hallucination 机制,模型反而给出了具有明确二级结构特征和可折返几何的 backbone。
这些由模型“自行生成”的初始构象并不是随机噪声,它们普遍呈现:
合理的距离矩阵分布
典型的 α/β 二级结构片段
甚至在某些任务中,出现与目标蛋白相互作用的初步界面几何
换言之,结构预测模型自发生成的“hallucinated backbone”可以被视为一种可用的模板空间。
在下图中(Fig 1D),这些初始骨架的表现非常直观:许多都已经具有紧致折叠,而非预期中的无序团块。
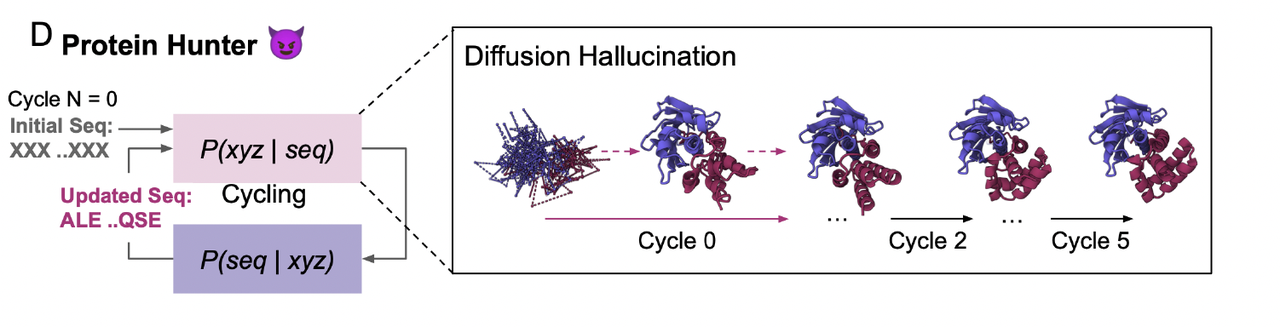
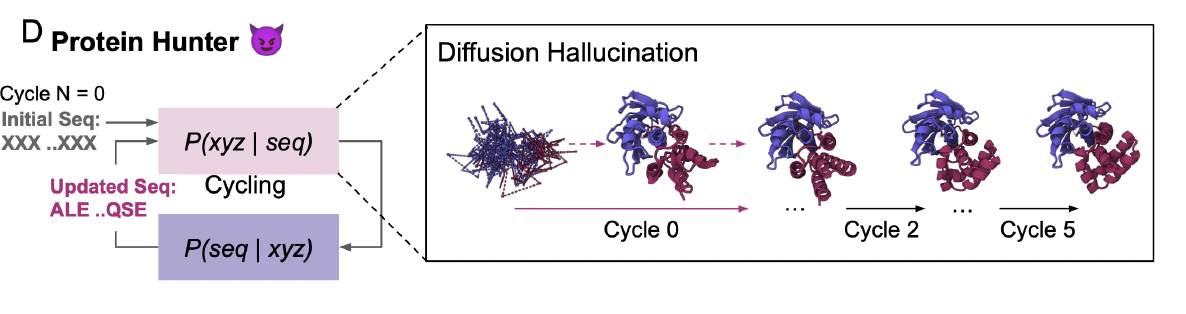
Protein Hunter:构象“幻觉”与序列优化之间的循环耦合
有了这些来自模型自身的“候选结构”,作者将其与高性能序列设计器(如 ProteinMPNN、LigandMPNN)形成闭环。整个流程可以抽象为:
构象 hallucination → 序列设计 → 构象再预测 → 再设计 → …
每一轮推理都在两个方向上推动模型向“真实可折返”的结构靠近:
结构预测模型负责提供折叠的“几何吸引力”
序列设计器则不断让序列更贴近该几何并提升局部物理性
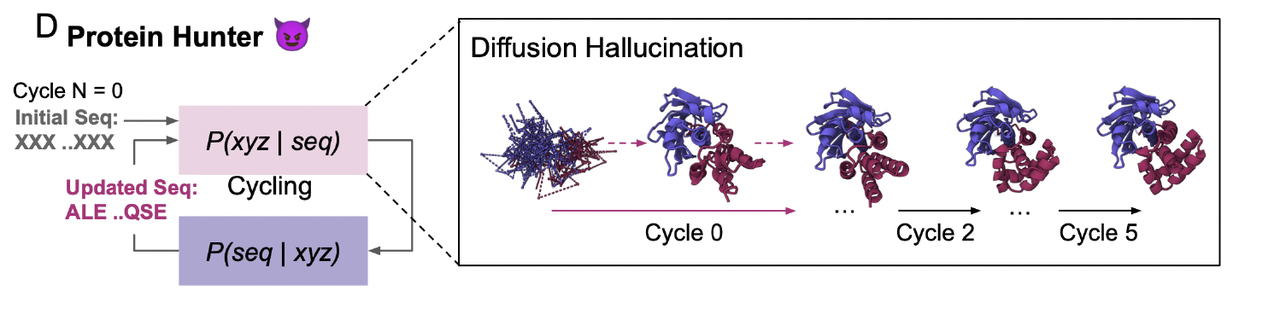
随着循环次数增加,原本松散的结构会逐步收紧,错误嵌入靶标内部的 loop 被自动修正,界面几何逐渐清晰。这种多轮迭代在论文第一张结果图(Fig 1D)中表现得十分明显:从 Cycle 0 到后续 cycles,构象的连贯性和界面完整度都有显著提升。
这一方法并不对结构预测模型本身做任何微调,而是完全基于“hallucination 行为的系统性规律”构建一个新的设计范式。
也正因此,作者将其称为 Protein Hunter:一种利用结构预测模型固有的构象 landscape,在其中“猎取”可设计骨架的通用策略。
二:从传统方法的局限出发——为什么我们需要新的设计范式?
蛋白设计在当前主要沿着两条思路演化:
一条是“先生成结构,再设计序列”的生成式路线;另一条则是“联合结构–序列空间直接优化”的梯度式路线。
这两类方法都有成功案例,但它们在大规模探索和通用性任务上都存在结构性瓶颈,这正是这篇工作试图突破的核心背景。
1. 生成式路线:结构虽能生成,但序列往往跟不上
代表性方法如 RFdiffusion,通过扩散模型直接生成 backbone,再用独立的序列设计器(通常是 MPNN 或 ESM-IF)去填充氨基酸。
这一思路的优点是:
生成速度快
易于控制骨架形态
对 binder、酶、环肽等任务有天然适配性
但其可扩展性被一个结构性问题限制:
生成的 backbone 未必是可折返的。
即使 backbone 在几何上看起来挺合理,后续序列设计步骤也可能难以找到能稳定折回这一结构的序列。
更直观地说,如果 backbone 本身落在了“不可折返区域”,那么序列优化的空间再大,也无法“扭”回到物理上合理的构象。
在论文示意图中,这类方法的流程被表现为:
结构生成是一条单向的扩散轨迹,而序列设计只是附加的一步。这种单次处理的结构,缺乏被反复修正的机会,因此对 backbone 的质量要求非常高。
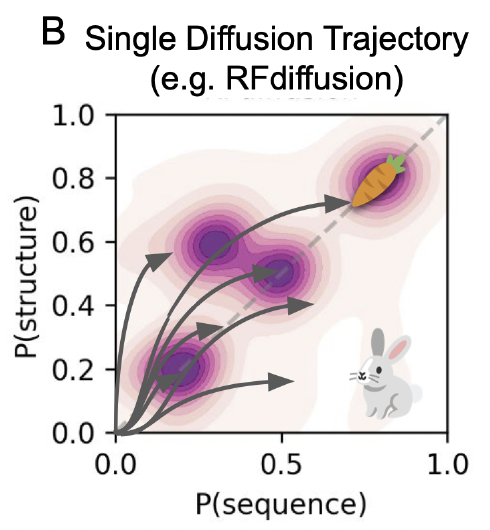
也就是说,这类方法常常需要生成大量候选,然后通过再预测、筛选、过滤来找出真正稳定的设计,成本不低。
2. 梯度式联合优化:理论完美,实际往往寸步难行
另一类方法,如 BindCraft、BoltzDesign 等,则直接在“结构–序列”的联合潜空间中做优化。
它们通常依赖:
结构预测模型的梯度
能量函数的梯度
序列类别分布的梯度
在理论上,这是最精确的方式,因为它能同时“推着结构走”和“推着序列走”,让二者高度耦合。
但在实际应用中问题非常突出:
优化路径容易陷入局部最优:特别是界面设计任务,优化一步错,后面就很难拉回来。
计算成本非常高:每一步优化都需要完整的结构前向–反向传播,对多聚体、大体系尤其昂贵。
对初始结构敏感:如果初始构象质量一般,梯度会把它推向离原任务越来越远的方向。
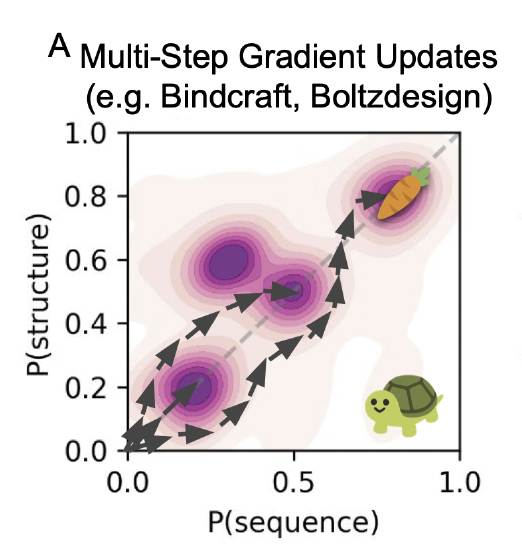
在上图里,这类方法的流程被体现为“一条长链式的迭代”:每一轮都伴随梯度传播和多次修正,反映了其计算复杂度与收敛困难。
3. 两条路线的共同瓶颈:都需要“可靠的初始折叠”
总结来看:
生成式路线:生成 backbone,但不一定可折返
优化式路线:能调整 backbone,但太贵、太慢、容易卡住
两者的共同点是:它们都缺乏一种“既能提供高质量结构,又能灵活迭代序列”的机制。
在理想情况下,我们希望:
起点的结构不要太差,最好天然具有一定折叠潜力
结构和序列的更新能来回耦合,而不是各走各路
不需要对模型重新训练,也不引入昂贵的梯度反向传播
还能适配不同目标(蛋白、小分子、DNA/RNA)
这是过去方法很难同时满足的一组条件。
4. Protein Hunter 的切入点:把“幻觉结构”变成 backbone 生成器
基于前面总结的限制,不难理解作者为什么会尝试“利用 hallucination 来做初始化”。
既然结构预测模型天然能:
从完全无信息的序列中构建合理骨架
通过多轮推理收敛到自洽构象
对界面几何、二级结构、折返可行性都有隐式 bias
那它实际上提供了第三种路径:
不再人工生成 backbone,也不强行对 backbone 做梯度优化——
而是让结构预测模型自己生成构象,然后通过循环过程不断洗炼它。
在论文的示意图中,这一思路表现为:
结构预测模型的推理本身就是“扩散式的重建轨迹”
只要与序列设计器形成闭环,结构–序列会在循环中自然收敛
这条路径既继承了生成式模型的速度,又具备优化式模型的稳定性
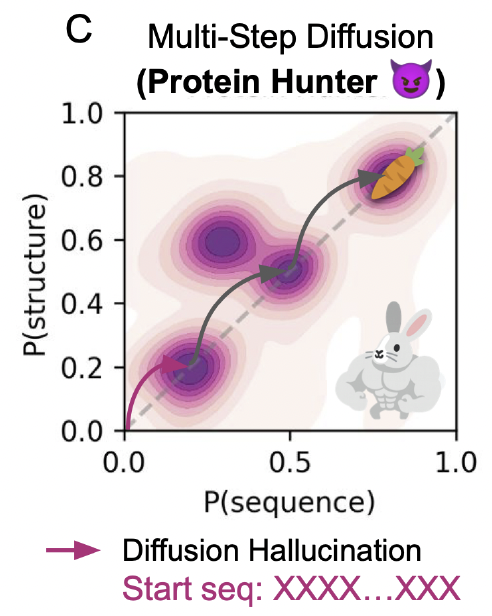
从此处开始,Protein Hunter 的方法逻辑就顺理成章:
既然模型会自发“做梦”,那我们就让它反复做梦,再反复修正,直到梦境成为一个真实可折返的蛋白结构。
三:Protein Hunter 的核心思路——让模型先构象化,再让序列去适配
传统方法常常在“怎么得到高质量 backbone”这一步上陷入瓶颈。而 Protein Hunter 的关键洞察是:结构预测模型本身就能生成 backbone,因此问题不再是“如何构造结构”,而是“如何利用模型的自然轨迹把结构洗炼得足够稳定”。
这一部分的方法逻辑可以拆成三个层次:
(1)如何从一条“全 X”序列获得构象;
(2)如何利用 MPNN 类模型让序列贴近结构;
(3)如何通过多轮循环逐步让二者相互收敛。
1. 从“全 X 序列”起步:让模型发挥其隐式几何偏好
Protein Hunter 的第一步是输入一条全由 X token 构成的序列。这个操作在直觉上看似极端,但在前一部分我们已经看到,AF3 风格模型在处理缺乏先验的序列时,会自然生成具有明显“天然蛋白特征”的骨架。
在论文的示意结构中,这些初始构象大多具备以下特征:
局部二级结构已经形成
远距离几何不再完全无序
全局折叠呈现紧致趋势
在与目标蛋白并排输入时,会出现初步的界面贴合
也就是说,模型的 hallucination 并非是随机的,而是落在一个充满结构偏好的能量景观上。
Protein Hunter 并不是违背模型的自然行为,而是顺着模型最擅长的那条路径往前推进。
这一点在示意图的“循环起点”处表现得很明显:第一轮的构象看似粗糙,但其整体轮廓已经“像一个蛋白了”。
2. 引入序列设计器:让序列逐轮贴向构象
仅靠模型 hallucination 得到的是 backbone,而 backbone 未必对应任何自然序列。因此,第二步的关键在于:
用一个序列设计模型,把 hallucinated 结构转写为具体氨基酸序列。
Protein Hunter 采用 ProteinMPNN 或 LigandMPNN 作为这一模块,原因在于它们具备:
强大的结构→序列映射能力
快速推理(数百至上千残基的序列也只需要几百毫秒)
能直接适配配体、界面几何等上下文
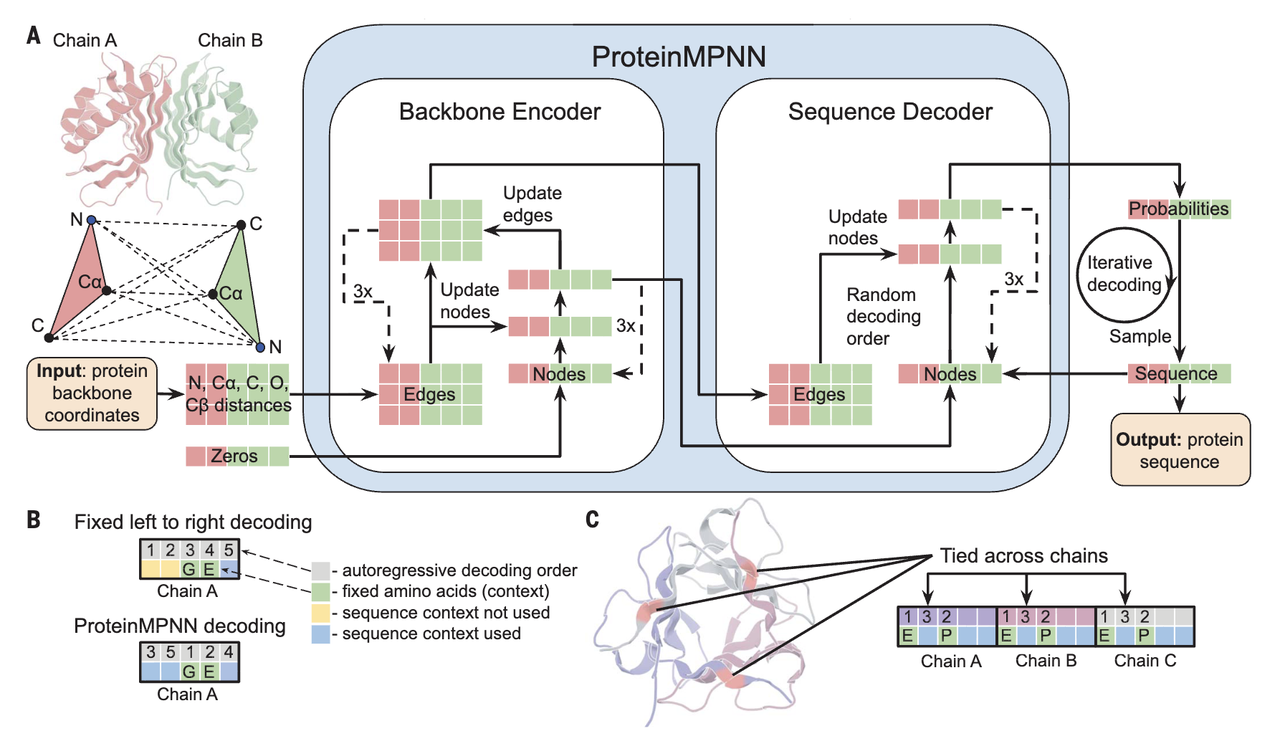
MPNN 的作用不是“强行把序列往目标方向推”,而是:
把结构预测模型“梦出来的几何”翻译成能维持该几何的序列。
这一步在方法图中对应的是“从 hallucinated backbone → 生成一个实际可折返序列”。
从机制上看,这相当于给几何结构加上“物理支撑”:模型的 hallucination 提供空间模板,而 MPNN 的序列填充为其增加稳定性。
3. 结构预测 ↔ 序列设计的闭环:构象逐轮被洗炼
Protein Hunter 最核心的创新并不是“让结构预测模型 hallucinate”,而是:
将 hallucination 与序列设计做成循环,把两者反复耦合,直到结构–序列共同收敛。
这一循环的机制非常类似“结构–序列的交替坐标下降(alternating minimization)”,但完全不依赖梯度,也不需要对模型微调。
流程可以概括为:
Step 1:模型 hallucinate 出一个 backbone
Step 2:MPNN 根据 backbone 设计序列
Step 3:把新序列再次输入结构预测模型 → 构象被重新折返
Step 4:得到新的 backbone,再用 MPNN 重新设计序列
重复 3–4 若干轮
每一轮,以下现象都会出现:
局部无序环(loop)逐渐减少
二级结构更趋于稳定
与目标蛋白的界面从“漂浮/穿模”变成贴合
pLDDT、pAE、ipTM 等指标会单调上升
序列中的不必要残基(例如初始时过量的 Ala)逐渐被替换
整体构象的可折返性显著提高
论文中的示例结构给出了非常清晰的视觉演化:从 Cycle 0 到 Cycle 5,构象的界面形状、骨架紧凑度、与目标的相对定位都趋于稳定。
换句话说,Protein Hunter 通过这种闭环机制,把结构预测模型的“隐式构象能量面”转变成一个可迭代搜索的设计空间。
4. 为什么这种循环有效?一个几何–序列共识的解释
从建模视角,循环有效的根源是:
结构预测模型的 hallucination 代表一种“几何偏好”
MPNN 代表一种“序列–结构一致性”偏好
两者的交替等价于:
在几何约束与序列约束两个能量面之间来回下降。
结构预测模型提供的几何偏好具有极强的规律性:其 hallucinated 结构往往处于“可折返的几何簇”附近。
MPNN 则更像是对这一几何的局部调节,让 backbone 获得合理的 amino-acid composition 和化学相容性。
循环次数越多,两者越可能在一个共同的局部最优点相遇。
这就是为什么结构会从粗糙走向精细,序列会从度量上不断变得更“物理”。
5. 该方法的一个重要特性:不修改模型,但能生成多样初始态
值得强调的是:
Protein Hunter 不需要微调结构预测模型
不需要额外训练 backbone 生成器
不需要引入人工定义的损失函数
不需要梯度
不需要外部能量函数
它的 backbone 多样性全部来自结构预测模型自身的 diffusion 轨迹。
不同的随机种子、不同初始噪声,都会让 hallucination 探索到不同的几何簇,使此方法天然具备结构多样性和构象探索能力。
这也使得后续所有应用(binder、小分子结合、环肽、核酸结合等)都可以统一地被纳入同一个框架。
第四部分:为什么可以从“全 X”开始?——幻觉结构的统计特性与 X token 的可靠性
Protein Hunter 的方法建立在一个关键前提之上:
结构预测模型对极度缺乏信息的序列仍然会生成具有天然蛋白统计特征的构象。
为了验证这一点,作者进行了系统性的“单氨基酸序列实验”,并通过对比天然蛋白结构库(CATH 数据库)的距离分布,全面评估模型 hallucination 的统计可靠性。
这一部分的结果强支撑了 X token 的选择,也使得“从全未知序列生成 backbone”成为可行的设计起点。
1. 单氨基酸序列实验:模型对无信息输入的响应具有强烈的“天然折叠偏好”
首先,作者构造了多个极端序列:
(H)_{120}, (N)_{120}, (A)_{120}, … 甚至是 (X)_{120},也就是整条序列中所有位置都相同,或全部为未知氨基酸。
对于常规理解而言,这些序列几乎不包含“可折叠性信息”,理论上模型应给出几乎无序的结构。
但实际情况恰恰相反:
这些序列经过 Boltz-2(以及其他 AF3-style 模型)推理之后,会生成有明确距离矩阵(distogram)模式的构象
很多构象呈现出明显的二级结构分布
其残基-残基距离分布与天然蛋白结构的统计规律高度相似
这一点在论文中的“幻觉 contact map”可视化里表达得极为直观:
尽管输入完全无信息,模型生成的 contact pattern 并没有呈现随机噪声,而是展示出典型的“局部接触密集、远距离接触稀疏”的天然分布格局。


换言之,模型的 hallucination 并不是无序的,而是带有天然蛋白进化统计学“先验”的。
2. 与 CATH 距离分布比较:X token 的 distogram 与天然蛋白最为接近
为了更量化地理解这种偏好,作者将 hallucinated 结构的距离矩阵与 CATH 数据库的残基-残基距离分布进行 Spearman 相关分析。
结果很清晰:
H、N 这类在天然蛋白中频繁出现的氨基酸,使 hallucination 的距离模式较接近天然统计
但 X token(未知氨基酸)反而是最接近天然统计分布的
多个模型(Boltz-2、Chai 等)的 distogram 结果都出现一致趋势
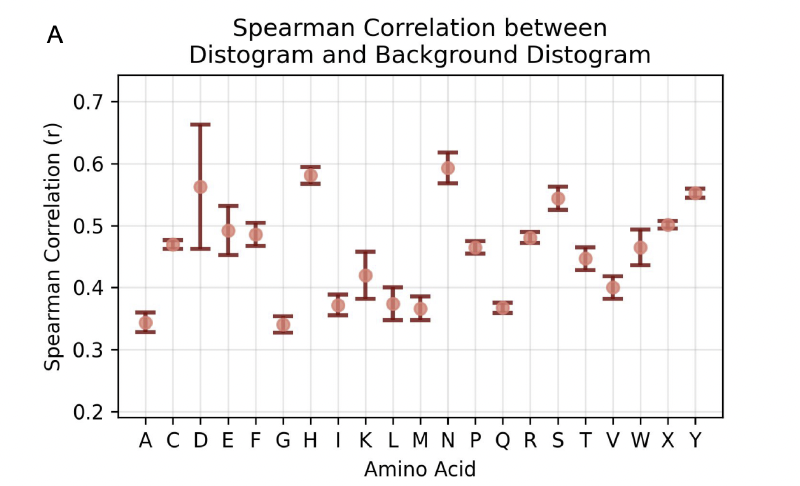
为什么 X token 会表现最好?
因为 X 不对模型施加任何位置偏好,它允许模型自由采样其内部的“几何吸引子”,因此产生的结构往往处于“天然构象簇”的中心附近。
在论文中,这种趋势不仅体现在 distogram,也体现在:
pLDDT 分布
refold RMSD
ipTM 分布
loop / helix / sheet 二级结构比例
这些指标一起说明:
X token 是一种能够诱导模型生成最稳定、最通用初始构象的输入形式。
3. 用 X、H、N 初始化的 binder 结构对比:X 的界面更稳、结构更收敛
作者进一步在具体的 binder 设计任务中比较了不同初始化方式的效果。
在一些展示任务中(例如靶向 CLDN1 的 binder),可以观察到:
用 X 初始化得到的 backbone 更紧凑
α-helix / β-sheet 的比例更合理
与目标蛋白的相对位置更稳定
ipTM 与 iPAE 指标整体更高
随循环次数上升,X 初始化更容易收敛
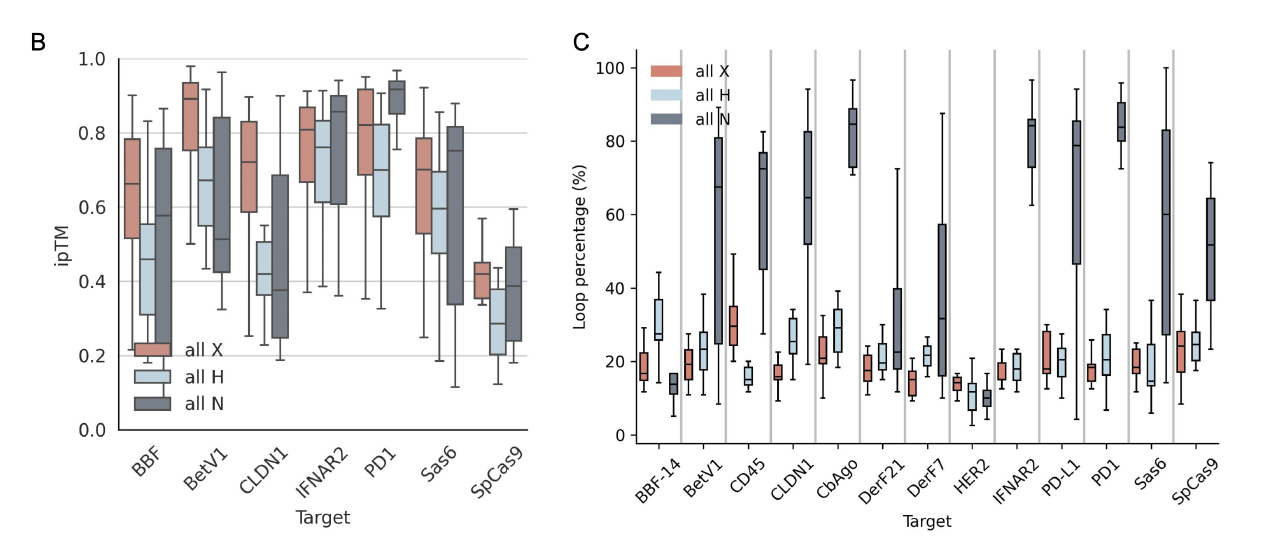
这些结果说明 X token 不只是从统计意义上“更像天然蛋白”,而且在设计任务中也能带来更优的界面几何、更快的循环收敛,以及更高的结构保真度。
因此,Protein Hunter 选择“从全 X 开始”并非直觉或巧合,而是经过系统分析后得出的结论:
X 是模型构象空间的最佳入口。
4. 多模型验证:幻觉结构在不同 AF3 风格模型中具有一致性
作者还比较了多个 AF3-style 模型(Boltz-2、Chai、自家 LF 模型等)的表现,发现:
这些模型在 hallucination 时,都能生成具备天然蛋白统计特征的 backbone
Boltz-2 在稳定性、pLDDT、refold RMSD 等指标上略优
但所有模型在“从 X 开始”时都能跑出可折返、具有二级结构的构象
说明幻觉结构不是单一模型的特性,而是整个 AF3-style 架构的共性行为
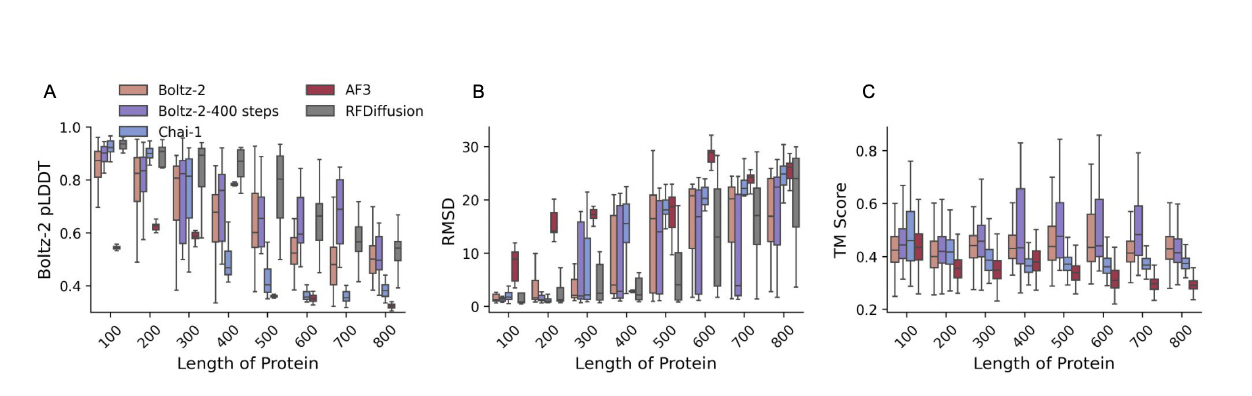
结合论文的可视化结构示例,不同模型 hallucinate 出来的骨架确实呈现跨模型一致性(例如偏 α-helix、偏紧致、界面结构自然)。
5. 小结:幻觉结构不是噪声,而是一种可利用的“几何先验”
经过这一大组实验,可以得出一个非常关键的结论:
AF3 风格模型的幻觉结构具有稳定统计规律,并且与天然蛋白的几何分布高度吻合。
这意味着:
从“全未知序列”生成 backbone 是一种可控、稳定的过程
使用 X token 作为初始化能高概率落入“可折返区域”
幻觉结构本身可作为设计流程的 backbone 候选
后续循环只是在这个几何簇内部进一步局部修正
这为 Protein Hunter 的后续一切能力(大蛋白生成、多聚体 binder、小分子结合、核酸结合、环肽等)奠定了坚实的理论基础。
第五部分:从粗糙到稳定——循环过程如何把幻觉结构“洗炼”成可折返构象
前一部分说明了:从一条全 X 序列出发,AF3 风格模型会生成具有天然统计特征的 backbone。
但要把这些“幻觉骨架”变成真正 可折返、可设计、可用于 binder 或 scaffold 的稳定结构,仍然需要结构–序列的反复耦合。
Protein Hunter 的循环机制不仅是概念上的“迭代”,它在结构质量、序列分布和几何一致性上都表现出非常系统性的改进。本节展示这一过程的核心证据。
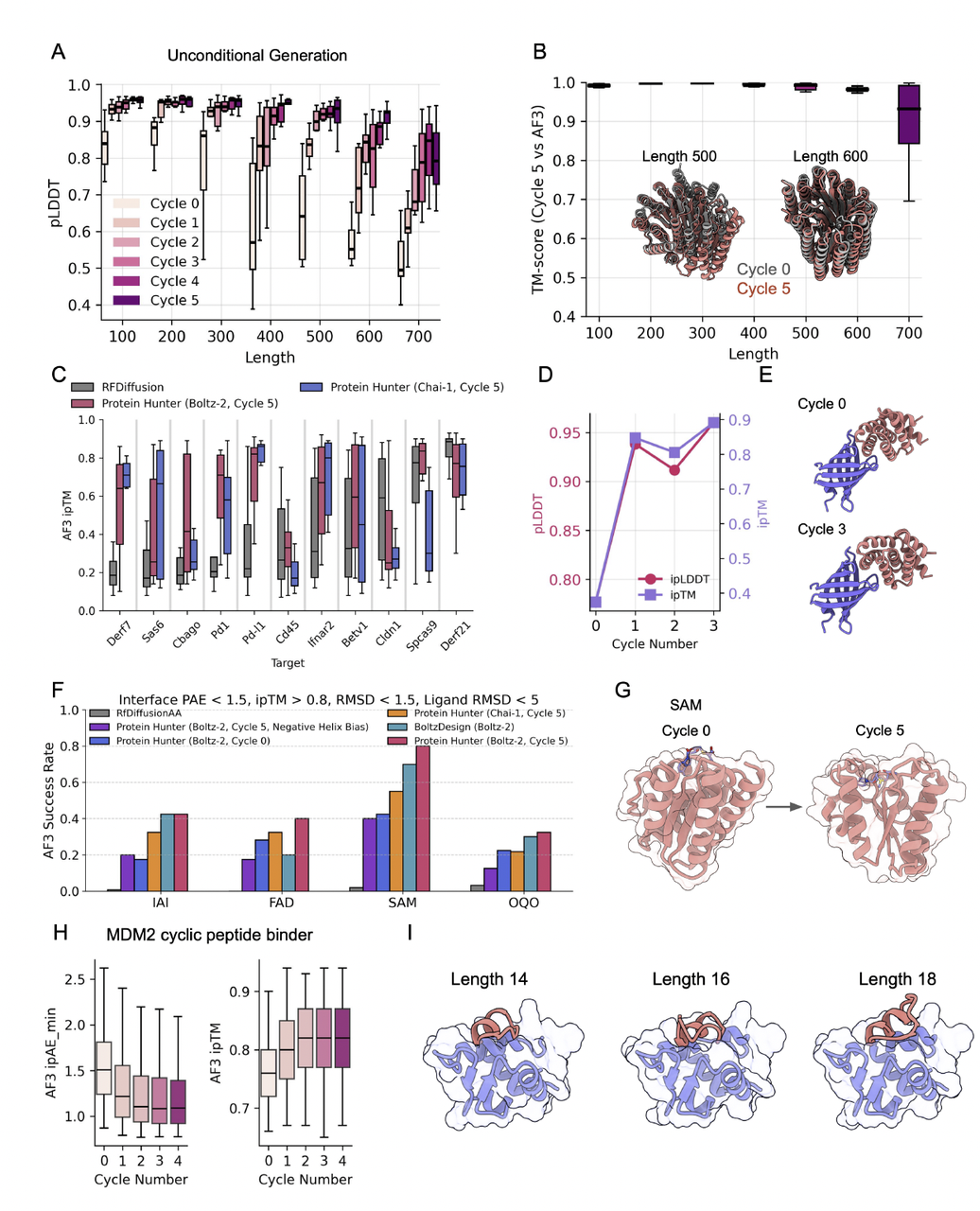
1. 单次 hallucination vs 多轮循环:长度越长,循环的优势越明显
作者首先做的是无条件蛋白折叠任务(unconditional protein design):
输入只有长度,不给任何模版、配体或结构信息,让模型自由“想象”一个折叠。
对于长度 100–700 aa 的蛋白,结果非常一致:
单次 hallucination 的 pLDDT 明显不足,尤其在 300 aa 以后下降明显
但只要经过 3–5 轮循环,pLDDT 会显著上升,并趋于稳定
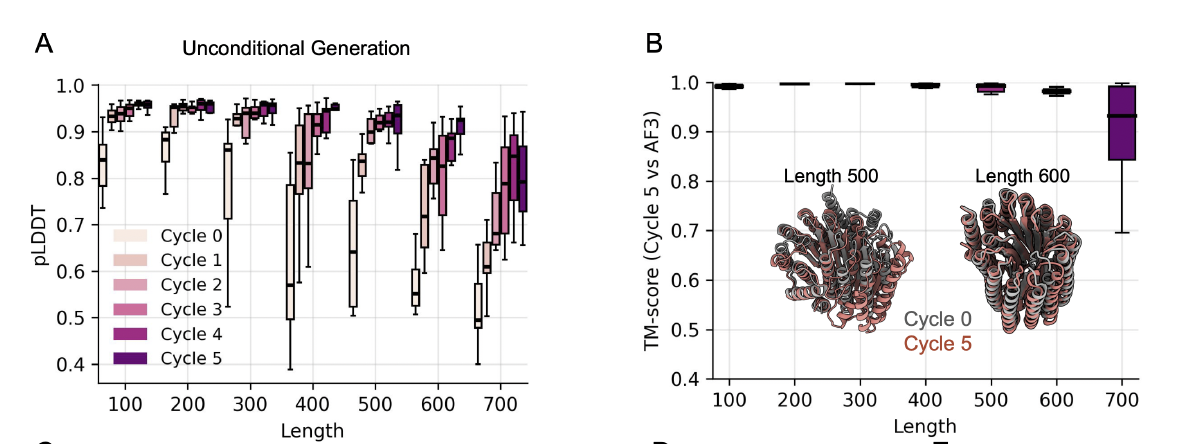
这说明一个重要事实:
幻觉结构在一次预测中虽然合理,但并未真正“自洽”;
而循环允许结构在模型的几何偏好中逐步“坐落到稳定区间”。
在上图右图里,你能看到一个 500 aa 的蛋白一开始结构很松散、远距离接触模糊,但随着循环,它变成了紧致的折叠,呈现出类似天然蛋白的 domain 组织。
2. Refolding 一致性:循环后的结构能被 AF3 再预测出来
判断一个模型生成的蛋白是否“真稳定”的标准是:
换模型或再预测时结构是否还能保持形状。
为此,作者让 AlphaFold3 再次预测循环后的结构(refolding)。
结果显示:
cycle 后的结构与 refold 结构的 TM-score 非常高(普遍 >0.7–0.8)
一级折叠与二级结构都能被独立模型“重现”
说明循环得到的构象确实落在 AF3 的“稳定簇”里,而不是模型偶然的随机输出
这一点是 Protein Hunter 能被用于真正蛋白设计的关键,因为 refold 稳定性意味着:
backbone 可折返
序列–结构组合在能量面上处于低谷
后续序列工程(如界面优化、功能化)具有可靠基础
3. 序列在循环中“去偏移化”:Ala 过量逐渐被清理掉
非常有意思的一点是:
如果让 MPNN 单次对 hallucinated backbone 做序列设计,序列往往会出现 过量的 Ala。这是因为:
初始 backbone 还未稳定
MPNN 倾向使用 Ala 来容忍几何不确定性(Ala 小、柔性高)
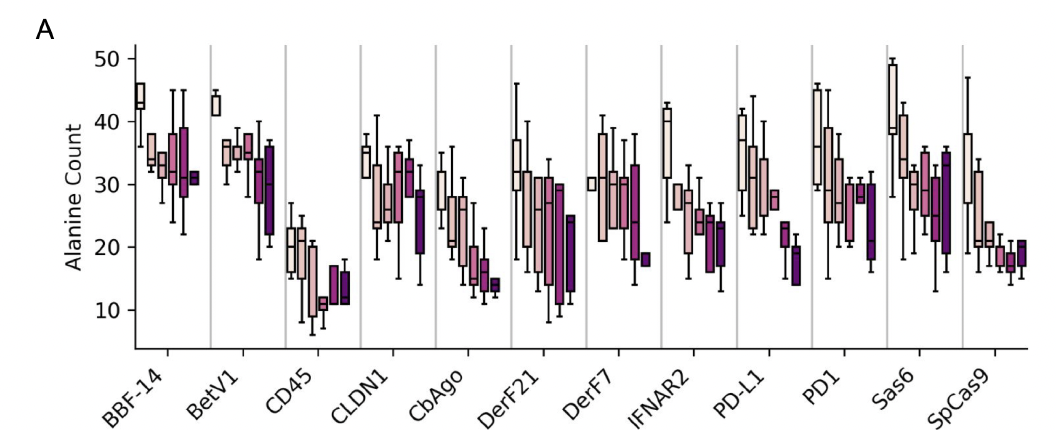
但在多轮循环中,序列组成会自动被“洗掉偏移”。
论文中的统计显示:
Ala 比例会持续下降
序列的氨基酸多样性逐渐恢复到接近天然分布
pLDDT 同步上升
各位置的熵降低(表示序列收敛)
一个直观的理解是:
当 backbone 逐渐变得更加确定,MPNN 不再需要依赖 Ala 来“缓冲结构噪声”,
因此可以更自由地选择更适合该几何环境的氨基酸。
这在构象–序列 co-design 的角度上极具意义:
循环不仅改善结构,也改善序列,使两者朝着同一能量簇共同收敛。
4. ipTM、界面几何与构象紧致性:循环让界面从“漂浮”变成“锁定”
在呈现给目标蛋白(如在 binder 任务中)时,单次 hallucination 的界面常常会出现:
“穿模”(binder 的 loop 伸进靶蛋白内部)
“漂浮”(binder 没有稳定地贴在界面上)
错位(界面角度不正确)
这些现象恰恰说明 hallucination 是“粗粒度的几何偏好”,但不一定满足精细界面需求。
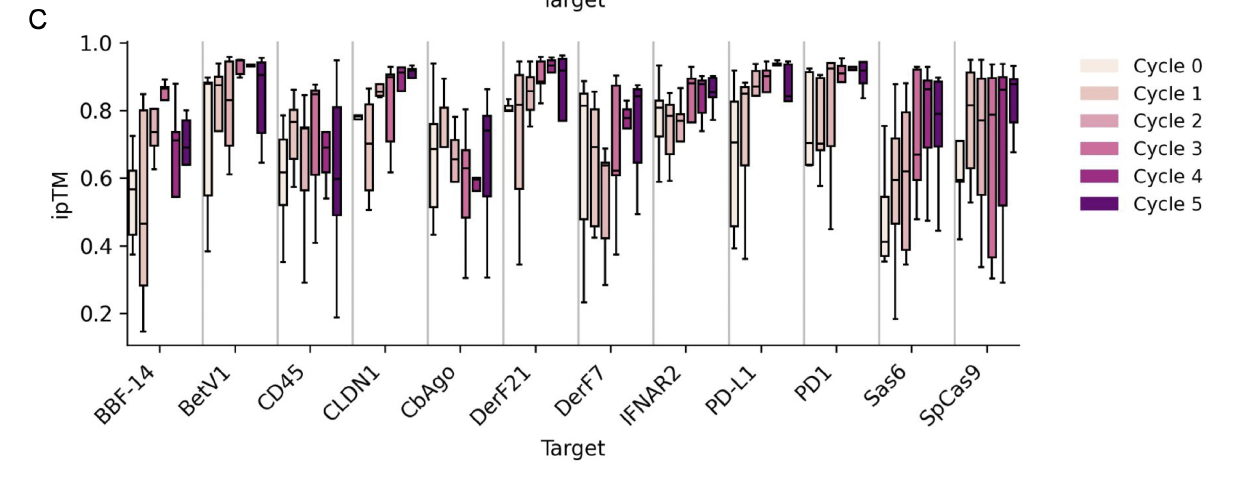
循环之后,界面呈现几个明显变化:
远距离相对位置开始稳定
binder 会自动转向更合理的朝向,而非“硬贴”在某个位置
ipTM 持续上升
接触面 becomes flattened and aligned
许多脱序 loop 会逐渐被“压平”或转变成 β-turn/helix
这说明循环不仅仅是在做“结构 refinement”,而是在使复合物的界面几何达到“预测模型认可的稳定构象”。
5. 小结:循环机制是 Protein Hunter 的真正动力
把这一节总结成一句话:
hallucination 提供粗粒度几何;
MPNN 提供序列一致性;
循环把两者锁定在一个共同的可折返构象簇中。
这一发现解释了 Protein Hunter 为什么能用极低的工程代价(不训练、不微调、不用梯度),实现跨任务的结构生成能力。
它也为下一部分(binder 设计)铺垫了一个重要结论:
只要 backbone 稳定,几乎任何界面类型都能被“套”到这套流程里。
第六部分:Protein–protein binder 设计——循环如何稳固界面,并在多数靶点上胜过传统方法
在有了稳定 backbone 的前提下,Protein Hunter 被进一步用于最核心的任务之一:蛋白–蛋白相互作用界面的 de novo binder 设计。这一部分的结果可以用几个关键点概括。
1. 在标准 binder benchmark 上,循环后的结构整体优于 RFdiffusion
作者测试了 11 个经典 binder 设计靶标。
比较对象包括:
RFdiffusion(代表传统生成式路线)
Protein Hunter(Boltz-2 / Chai 两个结构预测模型版本)
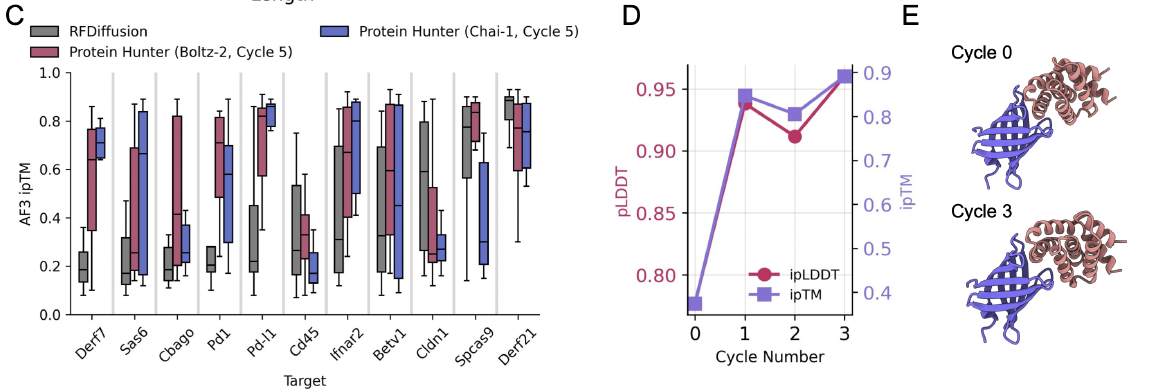
结果非常直接:
在 11 个任务中,Protein Hunter(Boltz-2 版本)有 9 个任务的 ipTM 更高
Chai 版本在多数任务上也表现更佳
界面 pAE(误差)显著更低
简而言之:
循环机制让界面逐步收敛到稳定构象,从而提高预测可信度。
RFdiffusion 虽然也能生成界面,但 backbone 质量不稳定,导致序列设计后的 refold 偏差更大。
2. 典型 case:从“穿模”到清晰界面
论文中的一个代表示例是 Bet v I(过敏原)的 binder。
这个例子说明循环的实际作用:
Cycle 0:binder loop 有明显穿模现象,甚至插入靶蛋白内部
Cycle 2:界面角度基本稳定,但二级结构仍不够稳定
Cycle 5:界面几何清晰、接触面连续、二级结构形成稳定骨架
这个例子表明:
幻觉结构提供大体方向;循环把界面从“模糊贴靠”修正为“锁定几何”。
这种界面收敛是传统一次性生成模型难以做到的。
3. 复杂体系:三聚体、四聚体的界面也能稳定贴合
更值得注意的是,这套方法不仅能处理单体或简单二聚体界面,也能处理复杂的多聚体目标。
其中最直观的是 TNF-α 三聚体:
初始 hallucination 时,binder 往往贴合得很松散
经过数轮循环后,binder 能稳定地贴附在 trimer 外侧的特定凹槽上
多个 binder 候选的 orientation 都一致,说明模型找到了可重复的几何吸引子
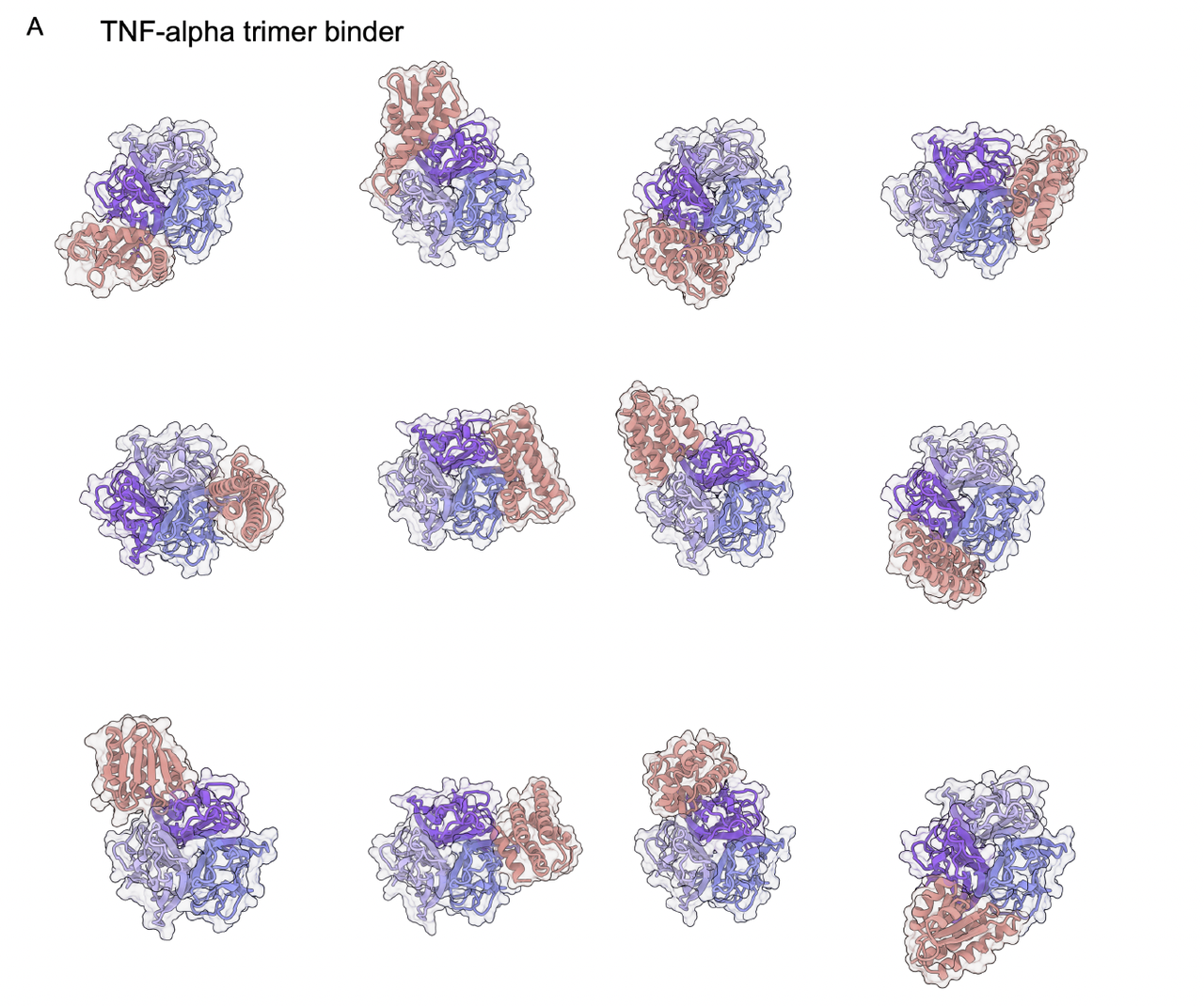
传统梯度式方法在这种多界面体系上通常需要巨大算力才能取得类似结果。
4. 循环的“界面几何强化”作用是核心差异点
综合来看,Protein Hunter 在 binder 设计任务的优势来自两点:
- 稳定 backbone 是前提
循环机制确保 backbone 本身落在可折返区域
界面几何在循环中逐步强化
ipTM 单调上升
误导性的穿模与漂浮现象会在后几轮被自动修正
这让 Protein Hunter 生成的界面具备:
更高对接稳定性
更明确的结合方向
更一致的界面形状
更高的模型置信度(pLDDT/pAE)
这是 RFdiffusion 或单次优化无法轻易获得的。
5. 小结:Protein Hunter 把“模型幻觉”转换为稳定的界面模板
一句话概括这一节内容:
在 binder 任务中,Protein Hunter 不是依赖一次性生成的骨架,而是利用循环不断强化界面几何,因此在多数靶点上显著优于传统路线。
这部分奠定了方法在蛋白–蛋白相互作用工程中的适用性,也为后续更复杂任务(小分子、核酸、环肽)打下了基础。
第七部分:跨分子类型的统一能力——小分子、环肽、DNA/RNA 都能被同一流程处理
Protein Hunter 的一个突出特点是:并未为特定任务设计专用模块,但依然能在多种截然不同的结合对象上取得稳定表现。
这背后的原因并不神秘——只要结构预测模型能够处理该分子类型,它的幻觉构象与循环机制就能自然延伸到所有配体。
1. 小分子结合
核心结论:循环能让 pocket 逐轮“收紧”,显著改善 ligand RMSD
传统的小分子 binder 设计通常需要:
预先定义口袋
或者引入物理能量项来约束小分子的相对位置
Protein Hunter 不需要这些做法。
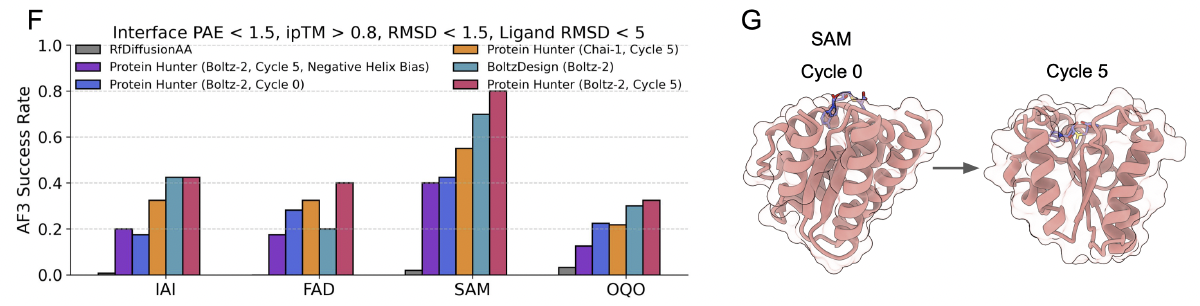
针对多个小分子任务(如 IAI、OQO):
Cycle 0:蛋白往往“绕过”小分子,口袋不完整
Cycle 1–3:小分子开始被包覆
Cycle 5:pocket 明显收紧,小分子位置稳定
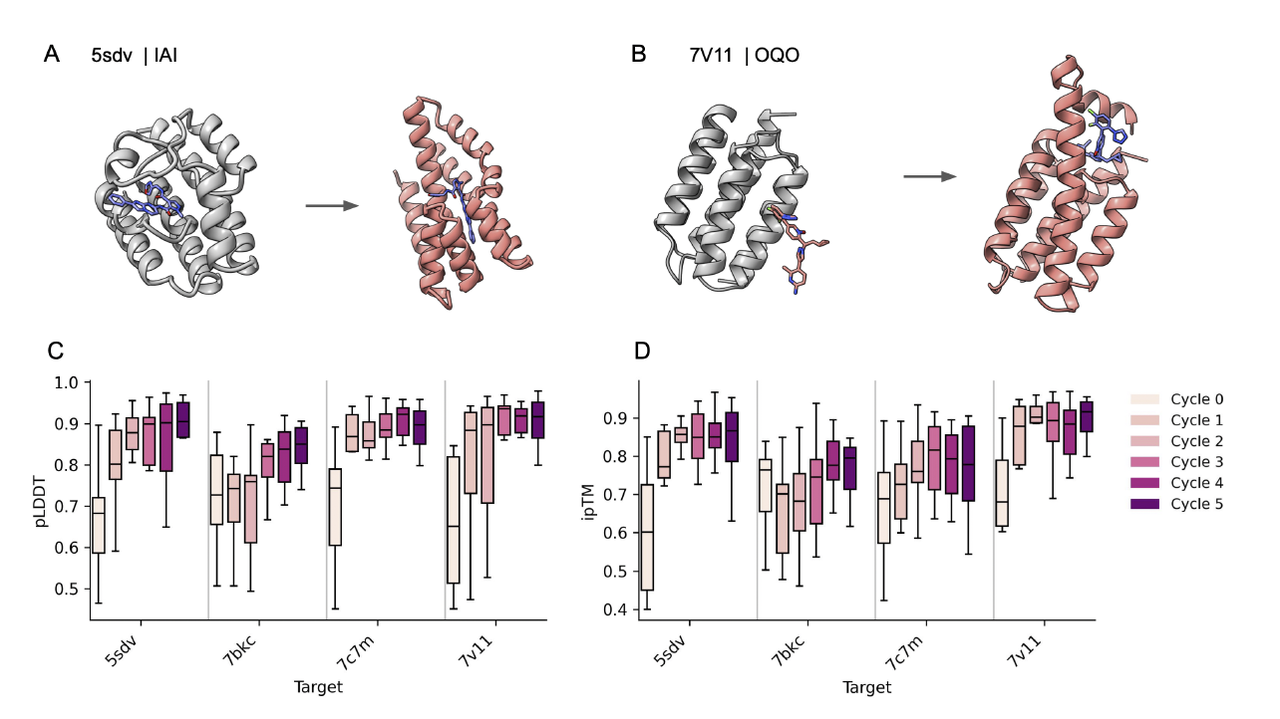
最终 ligand RMSD、interface pAE 都显著优于 RFdiffusion-AA 和 BoltzDesign2。
2. 环肽(macrocycle)结合
核心结论:环肽的构象非常难,但循环显著提升稳定性和界面几何
环肽是蛋白设计里最难的一类配体,原因是:
高度非线性
构象自由度大
传统序列–结构模型对环状结构保障不足
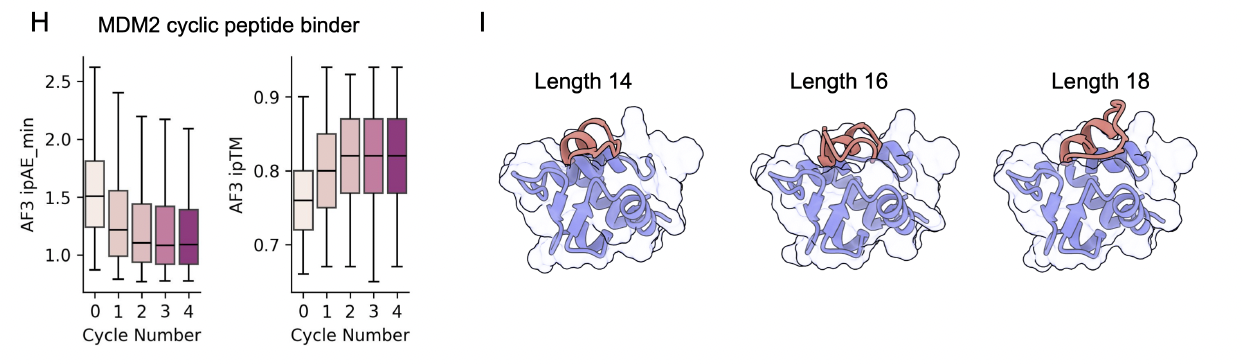
在 MDM2 环肽任务中:
iPAE(min) 在 cycle 中持续下降
ipTM 持续上升
14/16/18-mer 环肽都能形成完整界面
环肽自身的 backbone 变得更规则(β-turn、短 helix 明确成形)
这说明循环对 near-ring 的几何一致性提升特别明显。
3. DNA / RNA binder
核心结论:循环能让蛋白逐步贴合核酸沟槽,从漂浮变成锁定
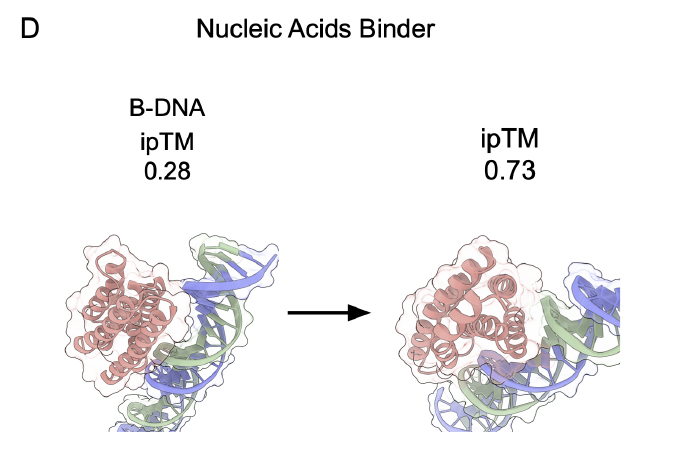
核酸 binder 设计的核心难点在于:
核酸负电荷密度高
groove geometry 相对固定
蛋白需要高度补形(shape complementarity)
在多个 DNA/RNA 任务中:
Cycle 0:蛋白通常“漂浮”在核酸旁边
Cycle 2–3:蛋白开始贴向 major/minor groove
Cycle 5:binding pose 固定且重复性好
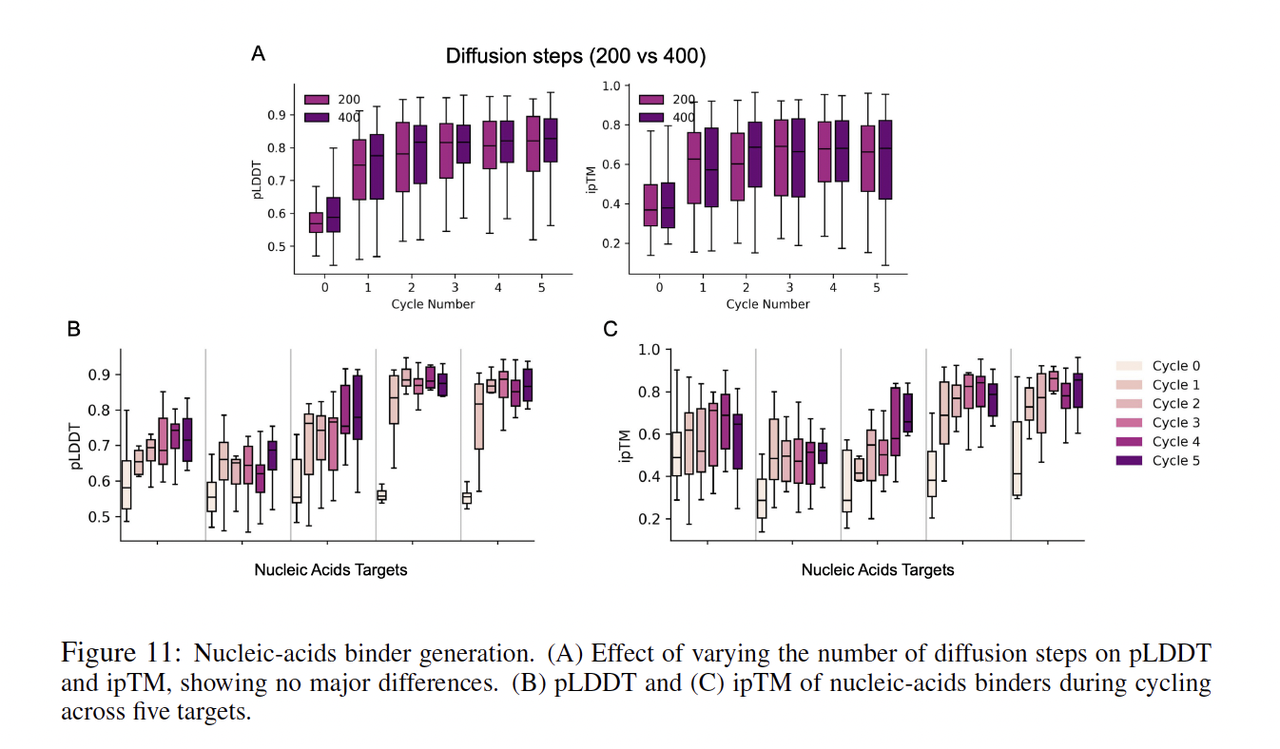
相应的:
ipTM 大幅提升
pLDDT 上升
接触面变得连续、规则
尤其在 B-DNA 的示例中,ipTM 能从 ~0.28 拉到 ~0.73,这属于质变。
4. 小结:为什么同一流程可以处理这么多类型?
原因非常简单:
AF3-style 模型已经能共同预测“蛋白 + 非蛋白分子”的结构;
Protein Hunter 只需要让模型在这个空间内反复收敛即可。
并不需要:
额外能量函数
特定类型的约束
针对性的 loss
只要结构预测模型能理解目标分子的几何,循环就能把 binder 结构洗炼到稳定状态。
这一点在论文中非常突出:所有任务——蛋白、小分子、环肽、RNA/DNA——都使用完全相同的流程。
第八部分:局部固定、全局补全——motif scaffold 与抗体 CDR 的统一重设计能力
除了从零开始的结构生成,Protein Hunter 在另一类高价值场景中也表现突出:
保留部分已知结构(如功能 motif、金属结合位点、抗体 framework),同时对其余部分进行 de novo 补全。
这是结构设计领域一直很难的任务,因为:
固定区域通常决定功能
自由区域必须“自发长出”能稳定支撑 motif 的骨架
传统方法往往需要专用 scaffold 生成器或高度调参
而 Protein Hunter 只用“幻觉 + 循环”就能完成同类任务。
1. Motif scaffold:heme-binding 功能片段的全局重建
任务设置非常具有代表性:
固定 3RGK 蛋白中的两个 heme-binding motif(离 heme <5 Å)
其余上百个氨基酸全部用 X token 进行初始化
让结构预测模型 hallucinate
再经过多轮循环让序列与结构共同收敛
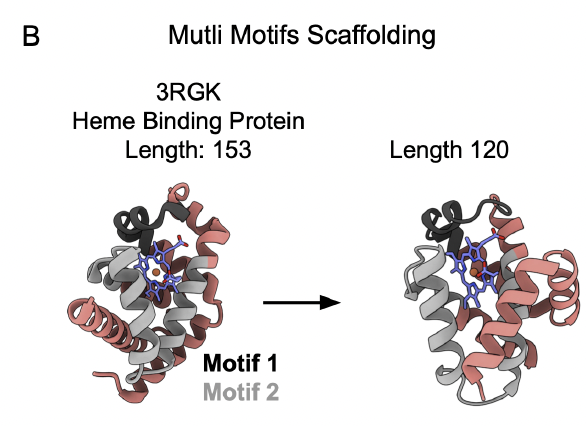
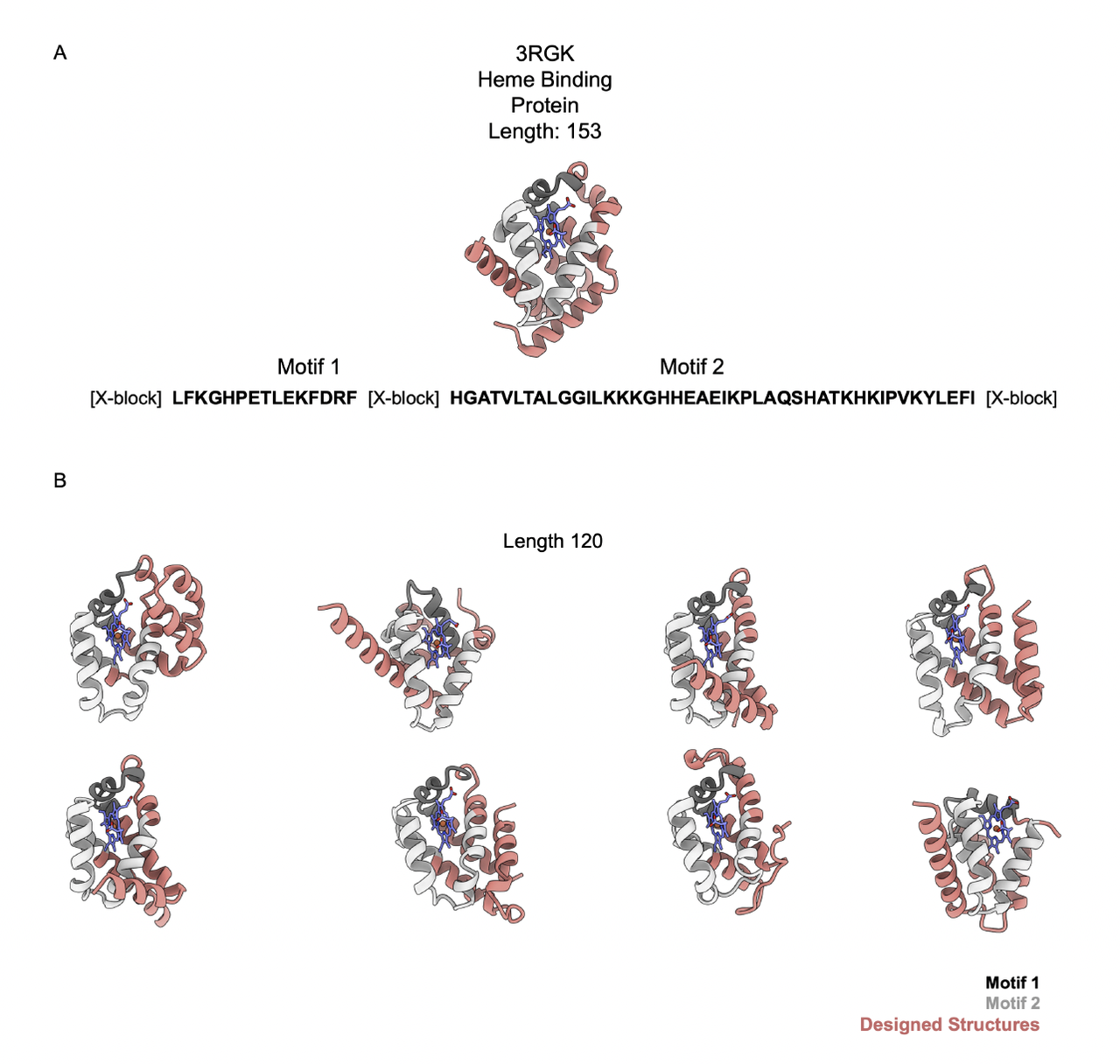
这些结果说明:
只要 motif 固定,模型会自动“长出”能够支撑 motif 的全局结构。
这与传统基于 scaffolding 的 pipeline 有根本不同,因为这里没有 backbone 生成器,只有“模型自己做梦 + 循环洗炼”。
2. 抗体 CDR 重设计:框架固定,CDR 区域 de novo 生成
抗体重设计是更典型的“局部固定 + 高度自由度”的任务。
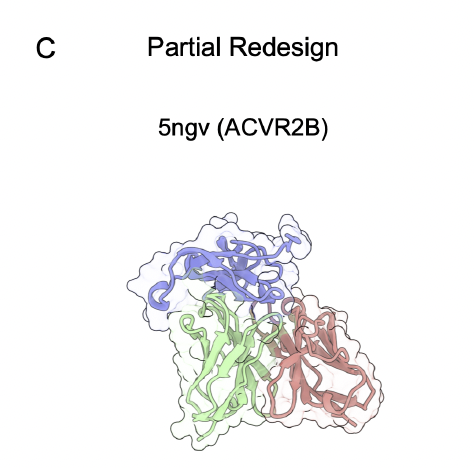
作者在两个体系上做了验证:
IgG 格式抗体(如 5NGV):固定 framework,仅重设计 CDR
VHH(纳米抗体)格式:固定 FR 区域,重设计特定 CDR
结果呈现一致趋势:
CDR 的初始 hallucination 常常偏松散、不贴合靶点
随循环推进,CDR loop 逐步收紧、定向化
与靶标的结合面变得连续、规则
ipTM 和界面 pAE 显著改善
多个独立设计的 CDR 朝同一 binding pose 收敛
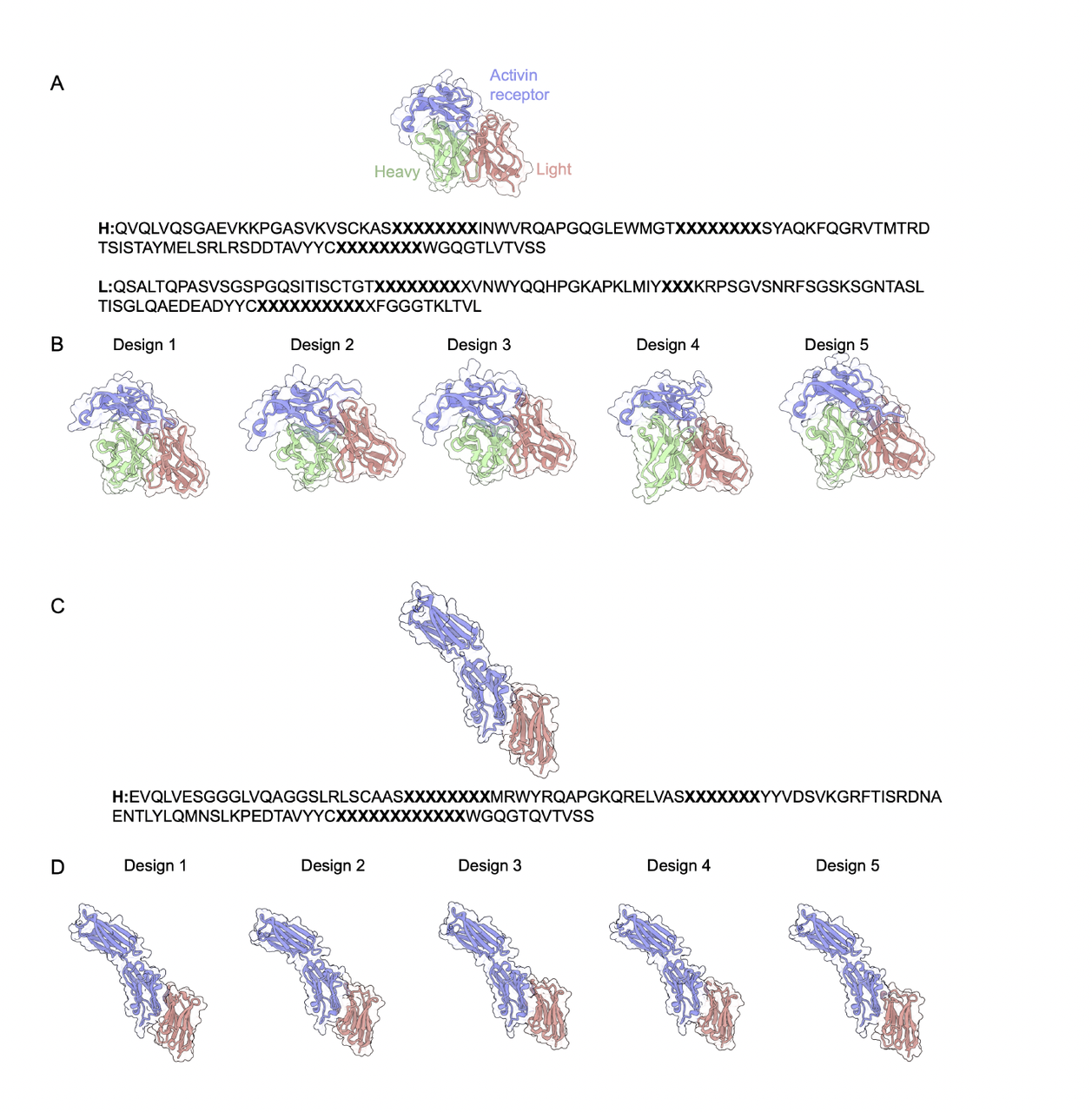
这说明 Protein Hunter 能够自动解决传统 CDR 设计里最棘手的问题:
如何同时生成可折返且能稳定对接的 CDR loop。
3. 为什么这一能力重要?
这类“部分固定—部分生成”的任务是生物设计最常见的实际需求:
抗体亲和力提升:固定 framework,只改 CDR
酶工程:保留催化基团,重建周围 pocket
受体工程:保留关键 motif,优化界面
药物递送 scaffold:固定结合片段,重建稳定折叠域
Protein Hunter 提供的优势在于:
不需要专用 scaffold 生成模型
不需要手动指定二级结构或骨架拓扑
不需要引入强约束的能量函数
完全由模型的几何偏好 + 序列一致性驱动收敛
这意味着它可以被直接嵌入各类功能工程任务中。
4. 小结:幻觉不仅能生成“整块结构”,还可以“补全结构”
一句话总结:
Protein Hunter 的 hallucination + 循环机制既能从零生成蛋白,也能以 motif 为核心向外“长出”合适的结构 scaffold,适用于 CDR、heme-pocket、酶活性位点等多种工程任务。
这为下一部分铺垫了一个重要前提:
不仅 backbone 风格可以控制,连折叠类型(helix-rich vs sheet-rich)也可以通过简单方式调整。
第九部分:结构风格可控性——用“负螺旋 bias”拓宽结构多样性
Protein Hunter 在大多数任务中的 backbone 都呈现出一个明显倾向:
模型偏爱生成 α-helix 丰富的结构。
这不是缺陷,而是 AF3 风格模型固有的几何偏好:
螺旋易折返、易稳定,因此在 hallucination 时自然更容易出现。
不过,在很多功能工程任务中(例如某些受体外域、免疫相关蛋白、β-rich scaffold),我们需要 更多 β-sheet。
为此作者提出一个极为简单但有效的技巧:在结构预测模型的 pair 特征上加入“负螺旋偏置(negative helix bias)”。
1. “负螺旋 bias”是什么?
作者修改的不是模型,也不是训练,而是:
在 Pairformer 输出的 pair features 上加一个简单的负偏置
这个偏置会抑制模型对螺旋距离几何的偏好(例如 i, i+4 接触模式)
偏置值从 0 调节到 -0.5,力度逐步增强
核心思想非常干净:
人为让“螺旋几何”稍微不那么被偏爱,使模型在构象空间更愿意往 β-sheet 区域走。
没有额外训练,没有能量函数,也不影响循环流程。
2. 效果非常直观:螺旋减少,β-sheet 大幅增加
论文的可视化结果非常清楚:
Fig. 12A:随着 bias 从 0 → -0.5
backbone 的螺旋逐渐减少
出现更大比例的 β-sheet 和 extended structure
Fig. 12B:不同 bias 下统计的 sheet fraction 分布
sheet fraction 显著提升
α-helix fraction 同时下降
二级结构的多样性被“拉开”
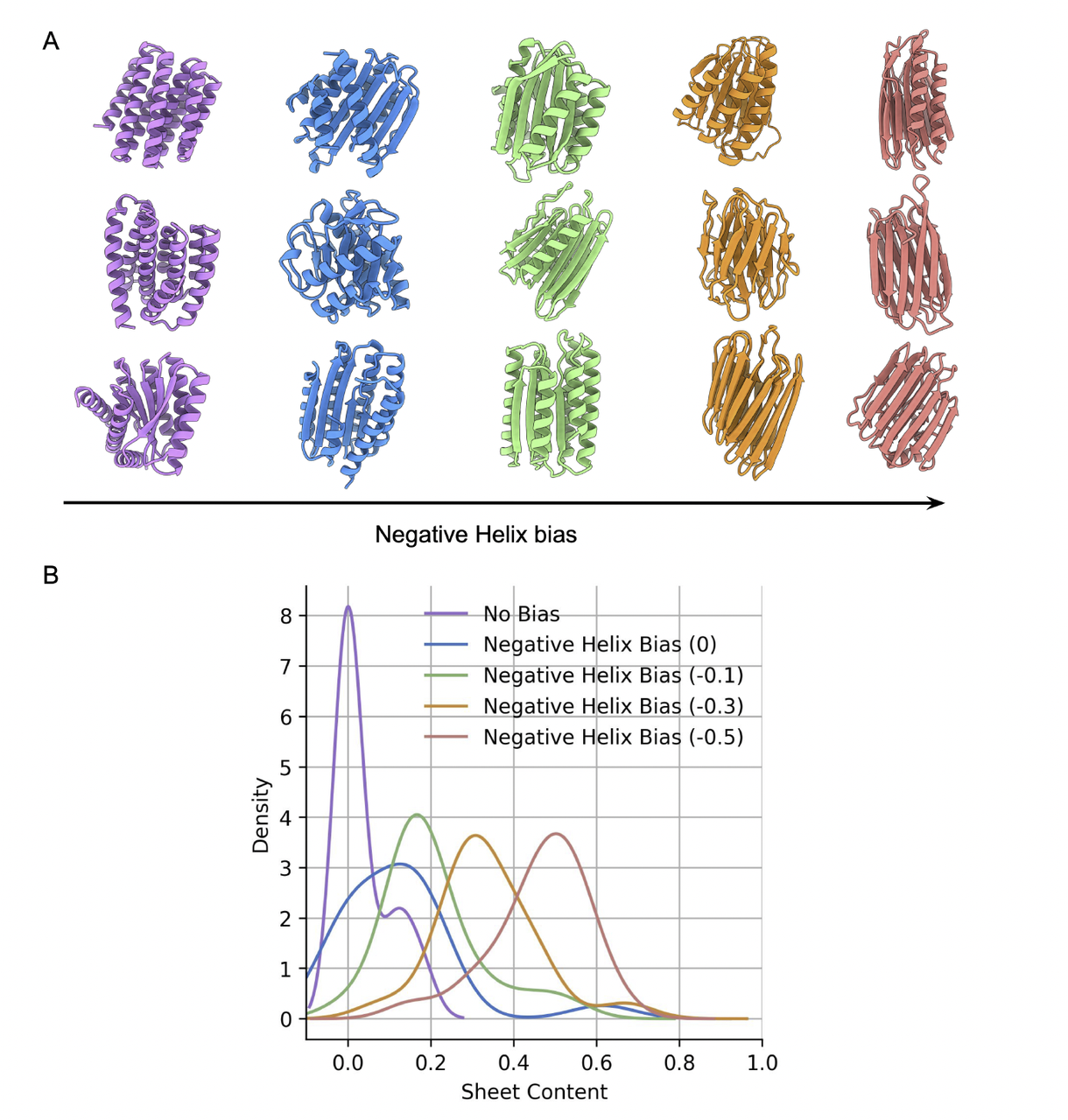
换句话说,这个 bias 成功把模型推离其默认的 helix-rich 吸引子。
3. 实际意义:风格可以被控制,而不限于“模型喜欢的折叠”
这一能力对于设计非常关键,因为:
螺旋-rich scaffold 虽然稳定,但不适合所有任务
很多天然受体(如免疫球蛋白域)本身是 β-sheet 为主
多样性更高的 backbone 能提供更广泛功能可能性
对 binder 任务来说,β-sheet 往往能提供更“平坦”的界面支撑
通过一个简单的偏置项就能控制整体折叠风格,使 Protein Hunter 的 backbone 库更广、更可调。
4. 小结:可控多样性是生成模型最重要的优势之一
一句话总结本节:
“负螺旋 bias”让 Protein Hunter 在保持稳定性的同时,显著拓宽了可生成折叠的风格范围,从 helix-rich 到 sheet-rich 都可以覆盖。
这为后续应用(特别是需要特定 scaffold 拓扑的蛋白工程任务)提供了更大自由度
第十部分:总结——把“模型幻觉”转化为通用结构生成引擎的设计范式
回顾全文,Protein Hunter 的贡献并不在于提出一个新的大模型,而在于发现并系统化利用了一个长期被忽视的事实:
AF3 风格结构预测模型的“幻觉能力”本身就构成了一种可控、可利用、可循环的结构生成机制。
传统蛋白设计的两条路线—生成式与优化式—各自的瓶颈非常清楚:
生成式方法 backbone 多样但不稳定,序列难以匹配
优化式方法序列–结构耦合强,但成本高、易陷局部最优
二者都难以做到“大规模生成 + 高稳定性 + 跨任务统一”
Protein Hunter 给出了第三条路线,解决了几个核心问题:
1. 让“幻觉结构”成为 backbone 生成器
从全 X 序列出发,结构预测模型自然会给出“带天然统计特征”的折叠。
这意味着 backbone 可以“自动长出来”,无需特定拓扑、无须能量函数、无需训练生成器。
2. 用结构–序列循环让构象稳定化、序列收敛化
MPNN 系列模型用序列一致性把幻觉 backbone“拉回物理世界”;结构预测模型再将该序列折返;如此循环数轮后,结构与序列共同落入一个稳定能量簇。
3. 同一流程拓展到所有类型的结合任务
Protein Hunter 不需要为不同配体编写专用模块:
蛋白–蛋白 binder
小分子 ligand
环肽
DNA / RNA
motif scaffold、抗体 CDR 局部重设计
只要结构预测模型能处理目标,循环就能使界面与骨架逐步稳定。
4. 结构风格可控(螺旋 vs β-sheet)
一个极简单的“负螺旋 bias”就能系统地改变模型偏好,使 backbone 风格从 helix-rich 扩展到 sheet-rich。
整体来说:
Protein Hunter 把结构预测模型从“评估器”变成了“生成器”,
并通过循环机制让这一生成过程变得稳定且可控。
这是蛋白设计领域中一个非常新的方向:利用现有预测模型的“内在几何偏好”去做设计,而不必从零训练大型生成模型。
延伸阅读
本文属于 AI4S文献 栏目。
返回 AI4S文献 → 去公众号阅读完整版 →