Nature Methods|从“蛋白自言自语”到“蛋白对话”:SWING 框架如何重写互作预测规则
今天要和大家分享一篇刚刚发表在 Nature Methods(2025 年 8 月)的重磅工作——Sliding Window Interaction Grammar (SWING)。
这是由匹兹堡大学团队提出的一种全新蛋白–蛋白/蛋白–肽段互作语言模型,它不仅在免疫学核心任务——pMHC 结合预测中表现出色,还展现了跨等位基因、跨 MHC 类别、跨物种的惊人泛化能力,甚至能识别氨基酸变异导致的特异性互作破坏。
为什么值得关注?
方法创新:它并没有依赖更深更复杂的网络,而是换了一个“视角”——先用生化差异把互作编码成一种“语言”,再让模型去学。
应用广度:从疫苗设计、疾病表位预测,到蛋白互作网络中的变异致病机制研究,都有直接落地价值。
泛化能力:在完全零样本的场景下(未见等位基因、跨 MHC 类别、跨物种),依然保持高精度,这对蛋白设计与免疫组学来说是非常罕见的能力。
接下来,我们就结合文章配图,一步步拆解 SWING 是如何从“氨基酸序列”读懂“蛋白对话”,并在多个领域实现全能预测的。
1. 引言:为什么需要新的“蛋白互作语言模型”
在蛋白质科学里,我们常说“结构决定功能”,但更准确的说法可能是“互作决定功能”(虽然结构决定互作)。绝大多数蛋白并不是孤立发挥作用,而是依赖与其他蛋白或肽段的精确结合。然而,即便有了 AlphaFold 这样的结构预测神器,现有的蛋白语言模型(protein language models, pLMs)在“读懂”蛋白互作上依然存在短板。
为什么?
答案如下图所示:
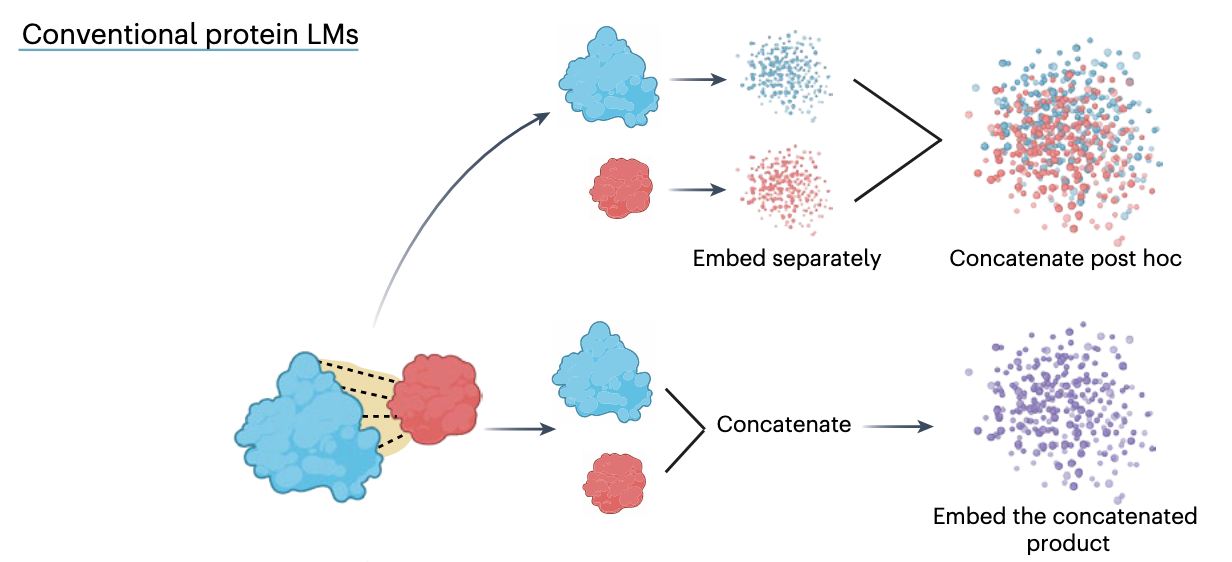
传统 pLM 通常会分别对两条蛋白序列做 embedding(序列编码),然后把结果拼在一起预测互作可能性。
问题是,这样的编码方式忽略了真正发生结合的局部残基(residue contact points),相当于把两篇文章直接合并,而不去关注它们交互的那几句对话。
结果是模型能捕捉到单条序列的特征,却对互作的“语法”理解不深。
这篇文章提出了一个全新的思路:
把蛋白互作当作一种语言,把每一次残基配对当作一个“单词”,用滑动窗口的方式读出整段“对话”。
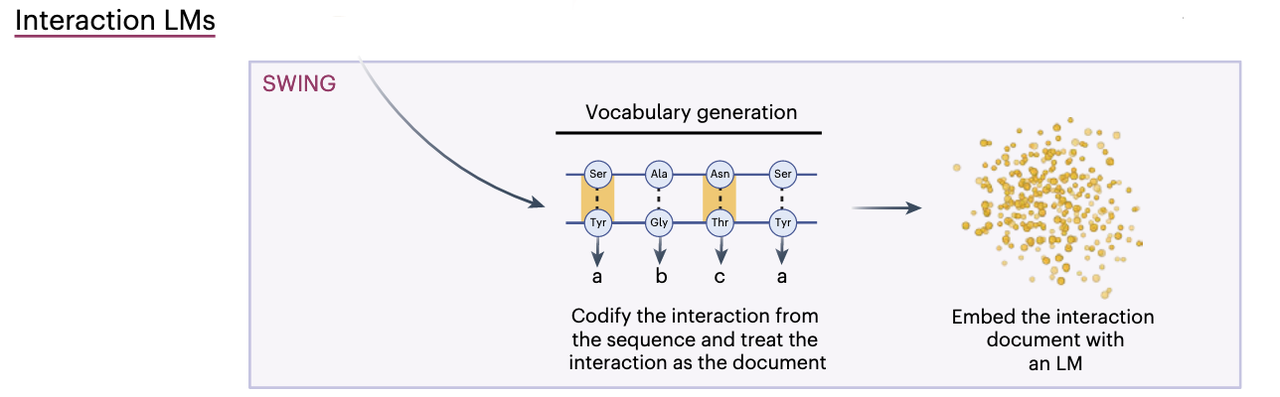
这个模型被作者命名为 Sliding Window Interaction Grammar(SWING)——字面意思就是“滑动窗口交互语法”。它的核心创新是直接在 embedding 前,把两条序列的配对信息转换成一个“生化词汇表”(interaction vocabulary),再交给语言模型去学习。
换句话说,如果说传统 pLM 是在学习“蛋白的自我独白”,那么 SWING 学的是“蛋白之间的对话脚本”。
2. SWING 核心机制拆解——从生化差异到交互语法
SWING 的核心思想是:在建模前,将两条互作序列的局部配对信息编码为“交互语言”,再让语言模型去学习其统计规律。 这一点在 Fig. 1b–d 中有清晰展示。
(1)滑动窗口匹配(Sliding Window)
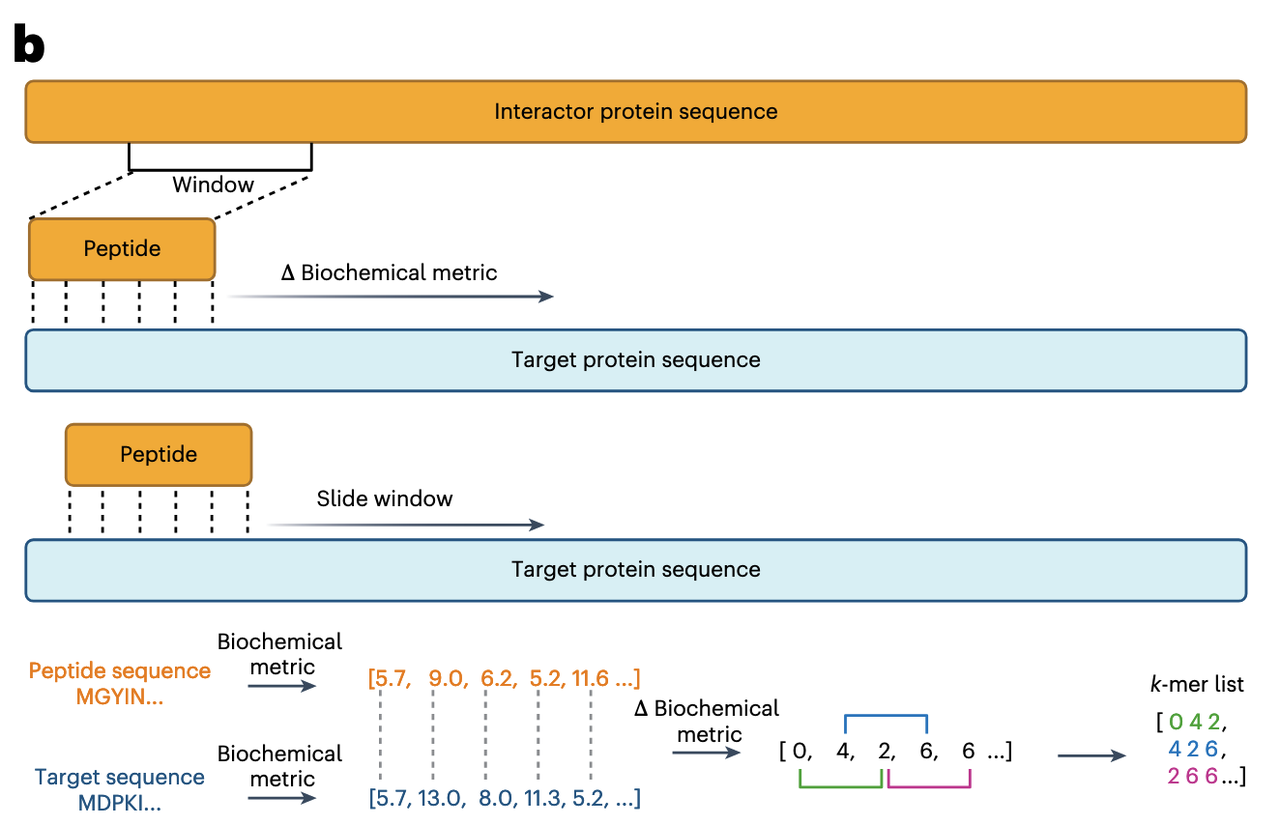
设定一个窗口长度 n(例如与肽段长度相同),将该窗口依次滑动匹配到互作伙伴的每一个可能位置。
在每个位置,对窗口内的氨基酸逐一配对,计算它们在某一生化尺度上的差异(如极性、疏水性等)。
得到的差异值是绝对值,经离散化(rounding + ordinal encoding)后,形成一个整数序列——这就是“交互语言”的基础符号。
(2)生成 k-mer 词汇(Vocabulary Generation)
将上述整数序列进一步分割成长度为 k 的重叠子序列(k-mers),每个 k-mer 对应一个“词(word)”,而整段滑动窗口配对结果就像是一篇“文档(document)”。
在 SWING 的语境中,每一次蛋白-蛋白或肽-蛋白互作就是一篇文档,k-mers 则是文档的词汇。
(3)Doc2Vec 嵌入(Embedding)
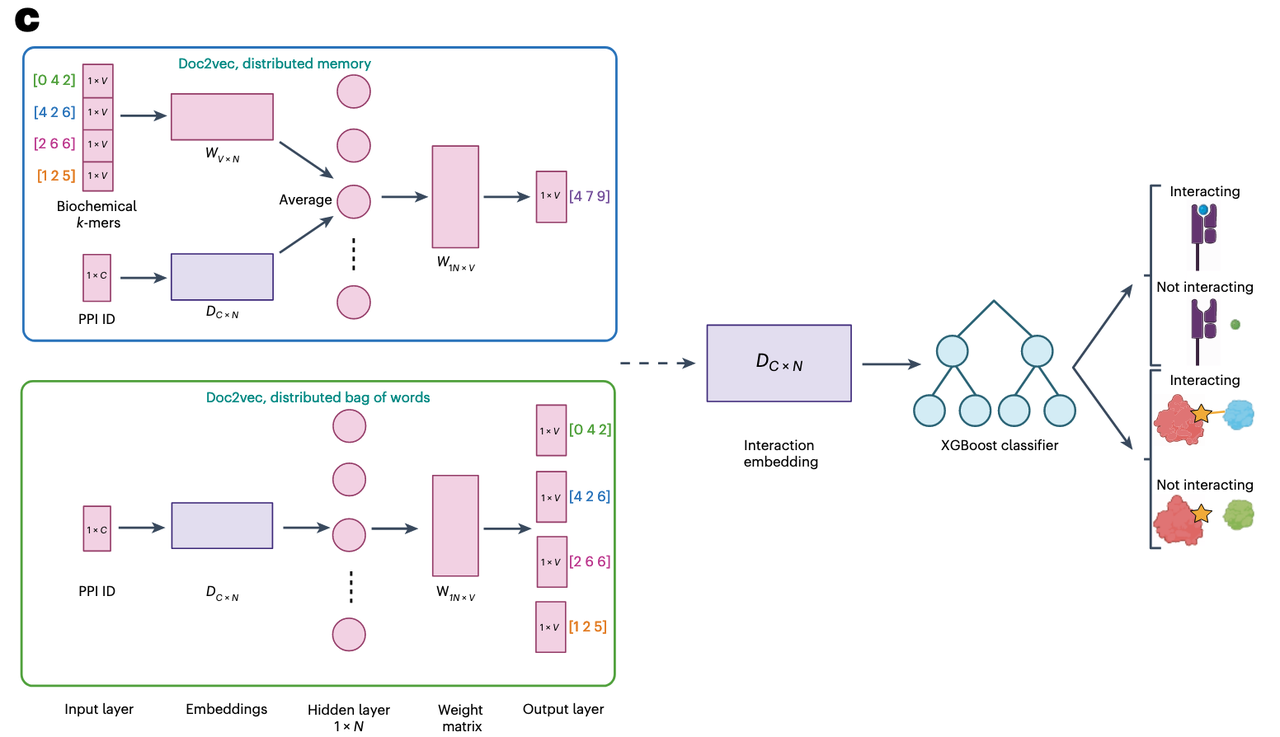
这些“交互文档”被输入到 Doc2Vec 架构中(Fig. 1c),生成固定维度的向量表示(interaction embedding)。
与直接用 pLM 对单条序列编码不同,这里的 embedding 是对配对关系本身进行建模,不再依赖对每条序列独立建模的结果。
(4)下游分类(Supervised Learning)
嵌入向量被输入到一个监督学习模型(文中使用 XGBoost)进行任务预测,如 pMHC 结合预测、变异致病机制解析等。
这种分离式架构的好处是,Doc2Vec 提供的交互嵌入具有高度的任务泛化能力,可以被任意分类器复用。
(5)关键创新(Fig. 1d)
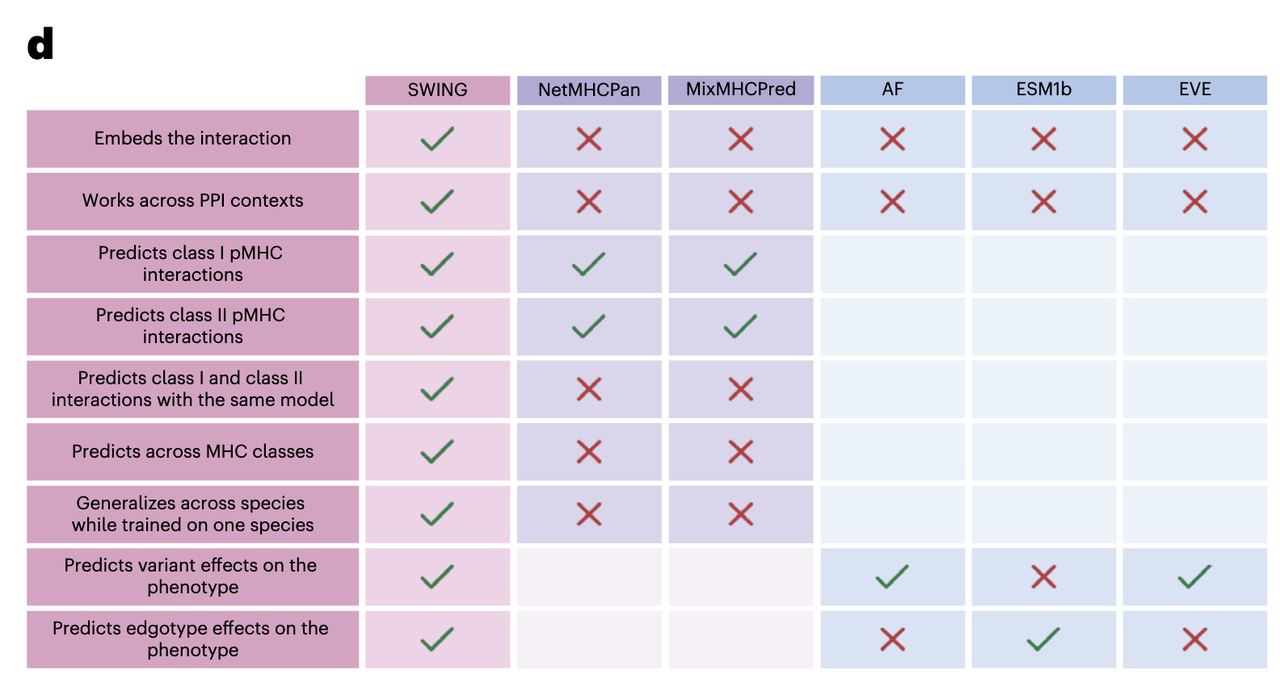
长度无关性(length-agnostic):不受序列长度限制,适用于跨物种和不同 MHC 类别。
跨上下文泛化(cross-context generalization):能在 Class I 与 Class II pMHC、甚至跨物种任务中保持高预测性能。
无依赖特定等位基因训练集(allele-independent):相比依赖等位基因特异性实验数据的方法,SWING 能对未见过的等位基因直接推断。
用更形象的比喻来说,传统方法是在“各说各话”后再猜测两个人会不会合作,而 SWING 则是直接分析他们在每一次眼神、每一句对话中的化学反应,从而精确建模互作模式。
3. pMHC 预测能力:从 Class I 到 Class II 再到跨类迁移
3.1 从已知到未知:泛化能力验证
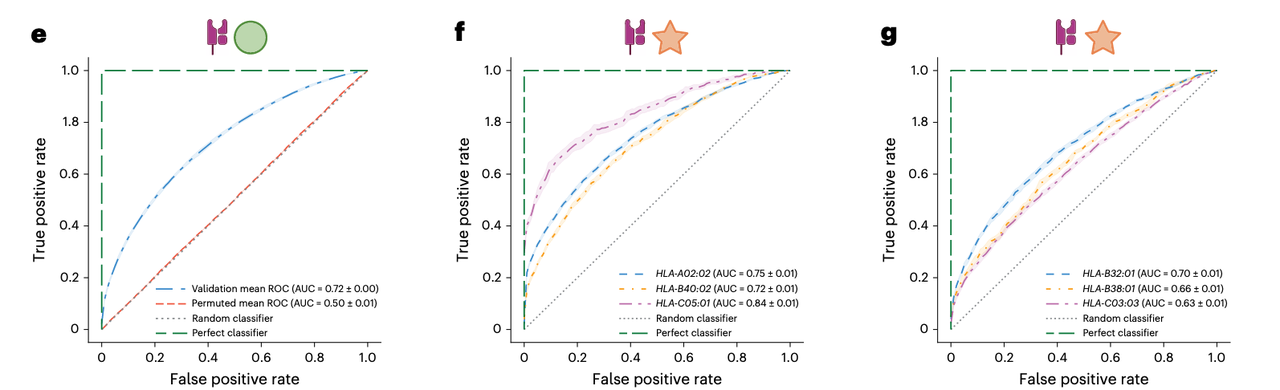
在 Fig. 2b–g 中,作者首先在 人类 MHC Class I 数据集上评估 SWING。
标准交叉验证(SCV):训练集与测试集共享部分等位基因,但 pMHC 配对不重叠。SWING 在这一设置下的 AUC 为 0.72(P < 0.001),显著高于随机水平(Fig. 2e)。
跨预测(Cross-prediction):训练集与测试集的等位基因完全不同,模拟真实“零样本”推断场景。SWING 依然在功能上差异较大的未见等位基因(如 HLA-A32:01、HLA-B38:01、HLA-C03:03)上取得 0.63–0.70 的 AUC(Fig. 2g)。
相似实验在 Class II 中重复(Fig. 2h–j),AUC 更高:
SCV 下为 0.90(P < 0.001)
未见 Class II 等位基因(DRB1_0102、DRB1_0404)上依旧保持 0.93–0.95
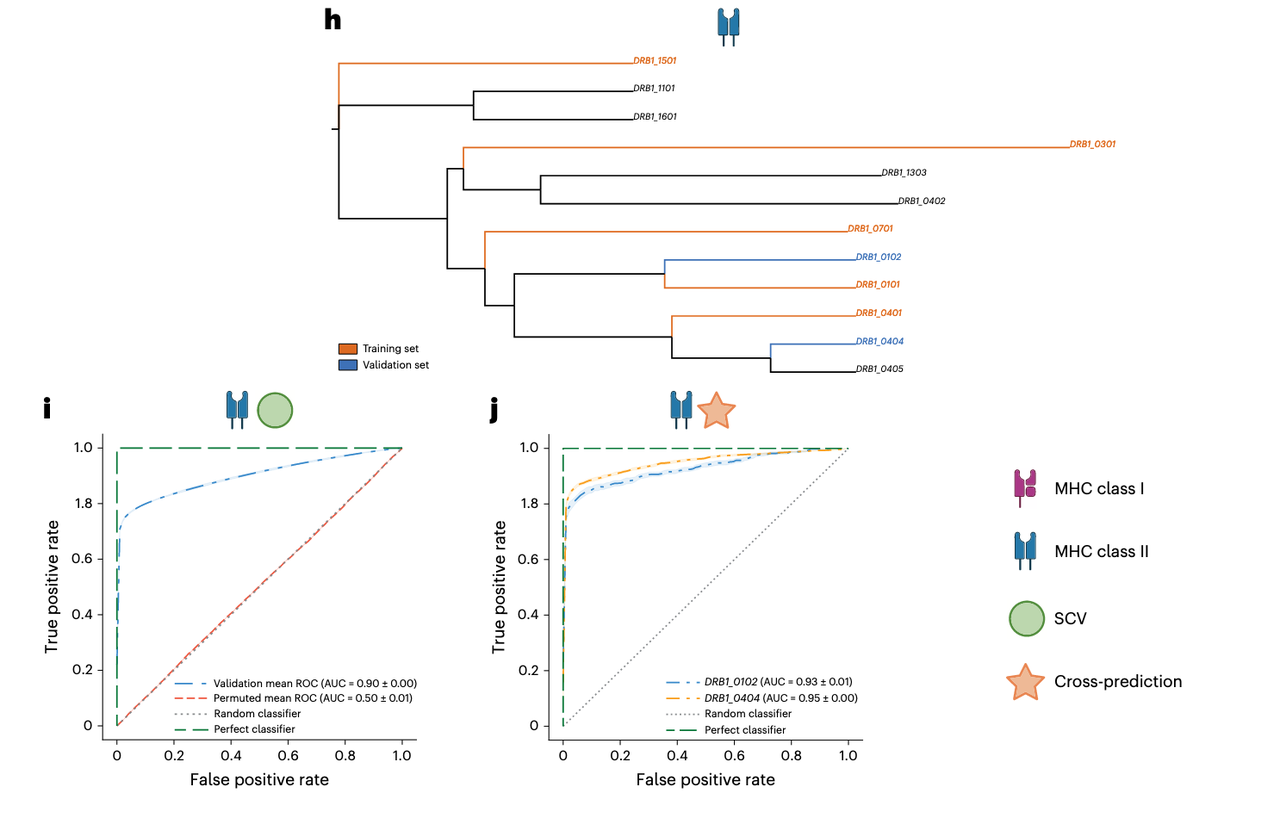
这说明 SWING 学到的是跨等位基因的“互作语法”,而不是死记某个特定 HLA 的结合模式。
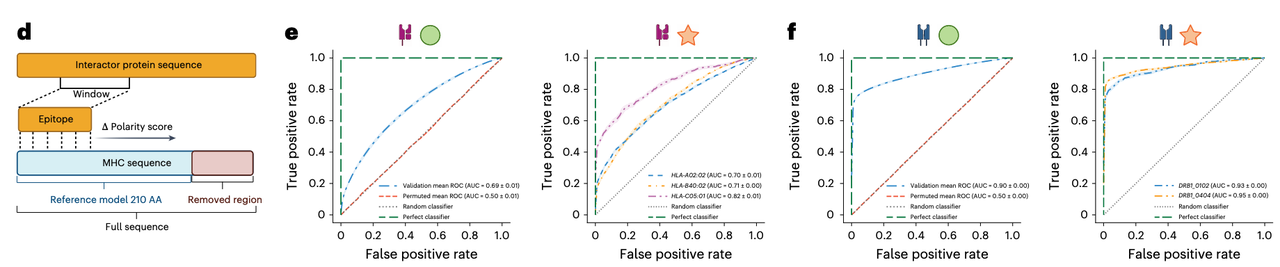
3.2 生化尺度的可替换性
在 Fig. 3a–c,作者将编码时的生化指标从极性(polarity)替换为疏水性(hydrophobicity),结果性能几乎不变(Class I: 0.72 → 0.72,Class II: 0.90 → 0.88)。
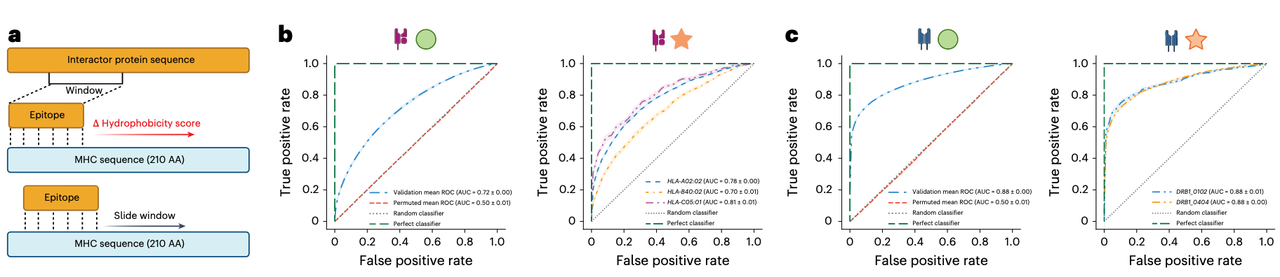
这表明 SWING 对生化指标具有鲁棒性,本质上是在学习相对生化差异的模式,而非依赖某个特定物理量。
3.3 免疫学机制的生物学解释
Fig. 3g–i 给出了一个重要的免疫学验证:
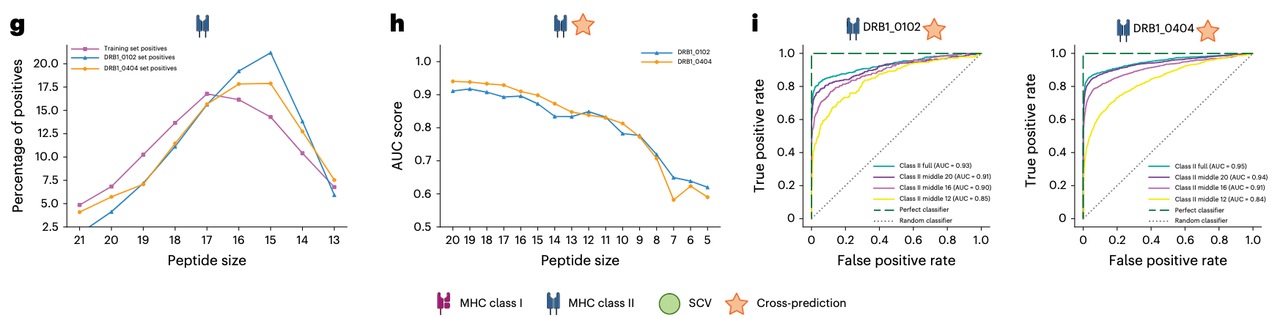
对于 Class II,肽段的 9-AA 核心决定结合特异性,其两端的延伸区域(flanking residues)也会影响结合稳定性。
当作者逐步截短肽段(从 21 AA 到 5 AA),AUC 在长度低于 9 AA 时出现明显断崖式下降(Fig. 3h)。
这说明 SWING 自动学会了核心区的决定性作用,而不是依赖人工定义的 motif。
这种“数据驱动学到免疫规则”的特性,使 SWING 有潜力在没有结构信息的情况下,推断未知等位基因或异常长度肽段的结合能力。
4. Zero-shot 与跨领域泛化
SWING 不仅能在单一 MHC 类别内部泛化,还展现了跨类别(Class I ↔ Class II)、**跨物种(人 ↔ 小鼠)**的零样本预测能力。这一部分在 Fig. 4a–i 中有系统展示。
4.1 Class I 模型预测 Class II(跨类别零样本)
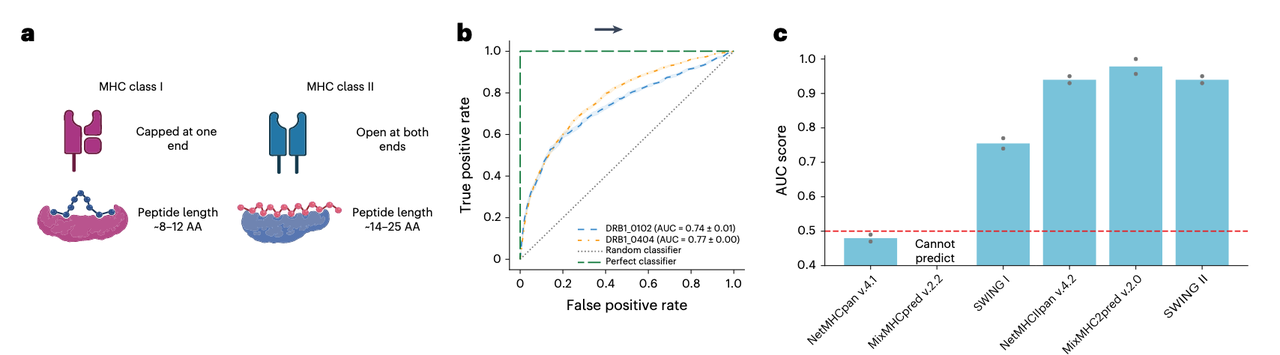
结构挑战:Class I 和 Class II MHC 的肽结合槽(binding groove)结构显著不同(Fig. 4a),前者两端封闭、限制肽长(8–12 AA),后者两端开放、允许更长肽段(14–25 AA)。
零样本结果:即便如此,只用 Class I 数据训练的 SWING 模型,在预测 Class II pMHC 时依然有 AUC = 0.74–0.77(Fig. 4b)。
对比:NetMHCpan(Class I 模型)在该任务上接近随机,而 SWING 保持稳定预测性能(Fig. 4c)。这说明 SWING 嵌入捕捉到的是互作共性,而非单一类别特征。
4.2 混合模型的联合预测能力
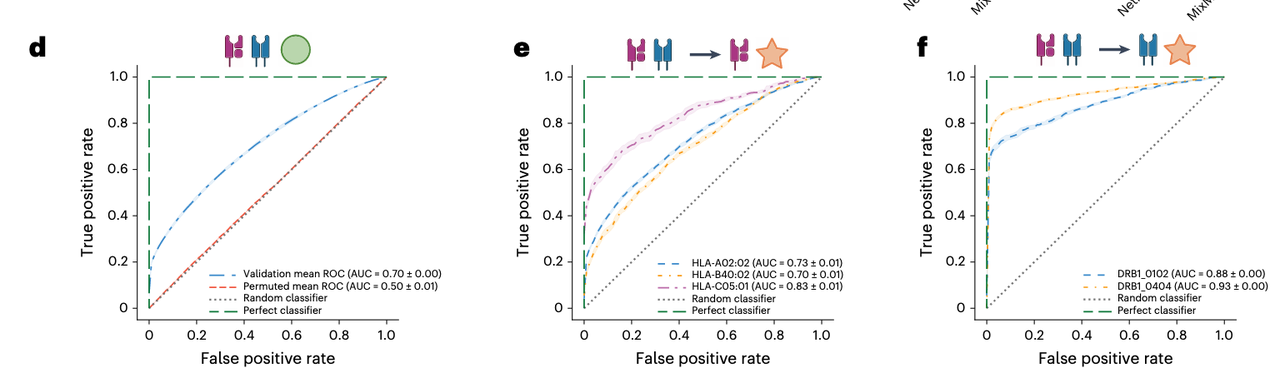
作者训练了一个 混合模型(Class I + Class II 数据同时训练),用于预测两类任务(Fig. 4d–f)。
结果显示,该模型在 Class I(AUC = 0.70–0.83)和 Class II(AUC = 0.88–0.93)上的表现,与单类别模型持平甚至更优。
这对于需要同时分析 CD8+(Class I)与 CD4+(Class II)免疫应答的研究场景尤为重要,例如综合性疫苗设计或免疫病理机制研究。
4.3 跨物种泛化(人类模型 → 小鼠)
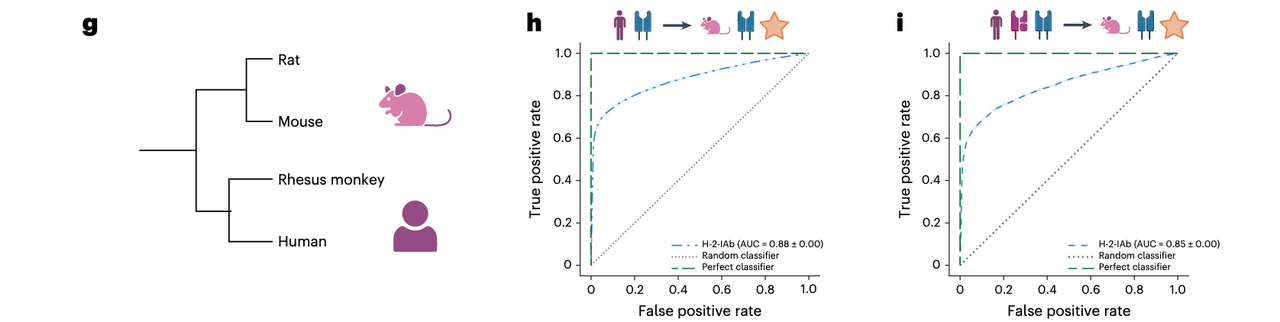
人和小鼠的 MHC(HLA vs. H-2)在序列和结构上均有显著差异(Fig. 4g),这对于大多数基于 motif 的方法来说是巨大挑战。
零样本结果:
人类 Class II 模型预测小鼠 Class II(H-2-IAb)时,AUC = 0.88(Fig. 4h)
混合模型预测同一任务,AUC = 0.85(Fig. 4i)
这一跨物种能力意味着,SWING 训练集可以完全基于人类数据,却能直接迁移到动物模型中进行免疫预测,大幅减少实验需求。
4.4 疾病相关应用案例:SLE 与 T1D
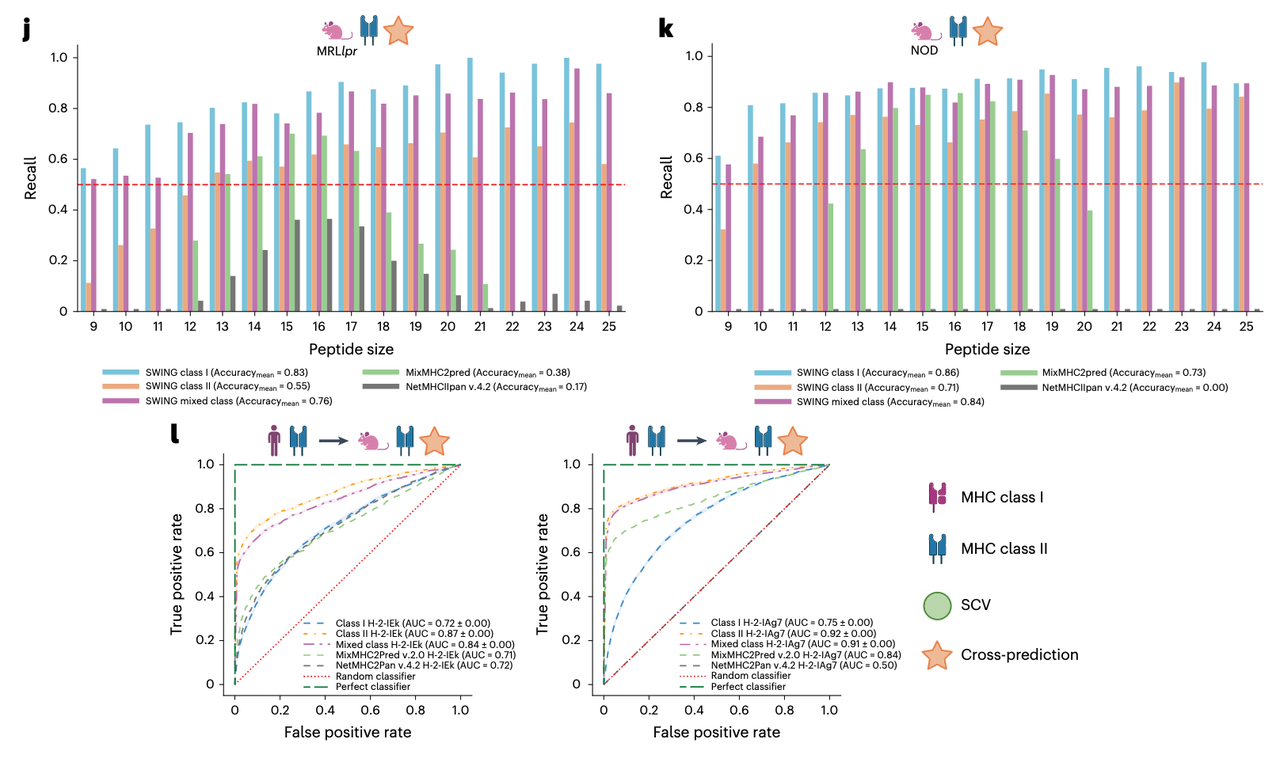
系统性红斑狼疮(SLE):小鼠 H-2-IEk 等位基因与狼疮性肾炎相关。SWING 在零样本条件下预测已知免疫渗透肽组(immunopeptidome),在各肽长上 recall 均优于 MixMHC2pred 和 NetMHCIIpan(Fig. 4j)。
1 型糖尿病(T1D):小鼠 H-2-IAg7(人类 HLA-DQ8 同源物)是高风险等位基因。SWING 未经该等位基因训练即可达到最高准确率(Fig. 4k),且对肽长无偏倚(Fig. 4l)。
这一部分的结果说明,SWING 不仅是一个“特定任务高性能”模型,更是一个具备跨类别、跨物种、跨等位基因的统一互作语言框架。它的嵌入向量包含了互作的生化本质,因此在缺乏针对性数据的情形下,依然能做出可靠预测。
5. 从免疫预测到变异致病机制解析
除了 pMHC 预测,SWING 还被扩展到预测氨基酸变异(missense mutation)是否破坏特定蛋白互作这一更普适的任务。这一功能在 Fig. 5a–l 中有详细展示。
5.1 现有方法的局限
主流的变异效应预测(Variant Effect Prediction, VEP)工具,如 AlphaMissense(AM)、EVE、ESM1b,更多关注蛋白整体功能或致病性(organism-level pathogenicity),而非互作层面的特异性破坏(interaction-specific disruption)。
这些方法在区分结合界面变异与非结合界面变异时表现良好(Fig. 5a),但在预测是否破坏特定 PPI时,性能显著下降(Fig. 5b–c)。
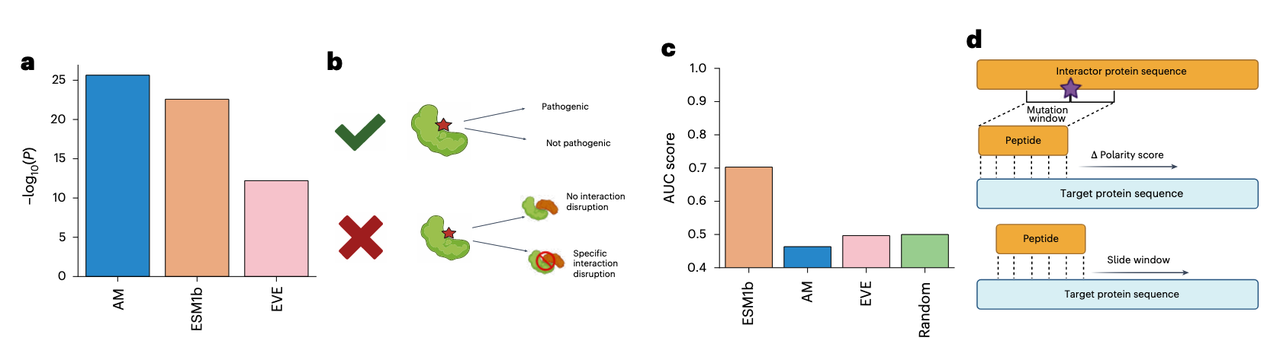
5.2 SWING 的适配与训练策略
在互作预测中,SWING 将滑动窗口对准包含变异的局部片段,与互作伙伴的序列进行生化差异编码(Fig. 5d)。
每个变异样本同时包含其野生型(wild-type)版本作为“未破坏互作”对照,从而让模型学习破坏性与非破坏性模式的差异。
5.3 性能评估
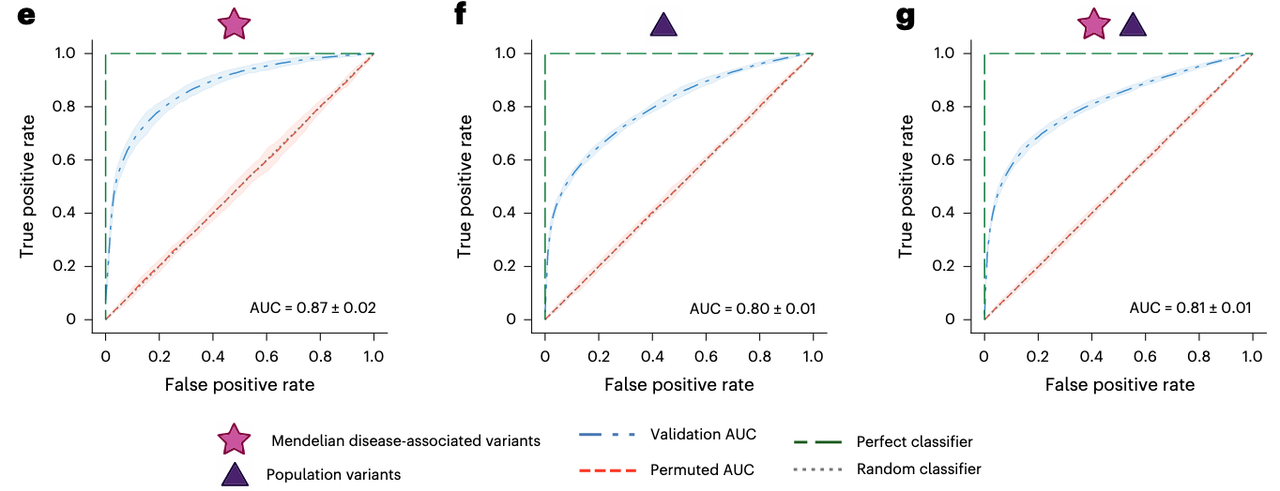
孟德尔病相关变异(Mendelian mutations):AUC = 0.87(P < 0.0001)(Fig. 5e)
人群常见与罕见变异(population variants):AUC = 0.80(P < 0.0001)(Fig. 5f)
混合数据集(来源无关):AUC = 0.81(P < 0.0001)(Fig. 5g)
模型在**跨序列簇(cluster hold-out)**验证中依旧保持 AUC ~0.71(Fig. 5i),说明没有过拟合到特定序列背景。
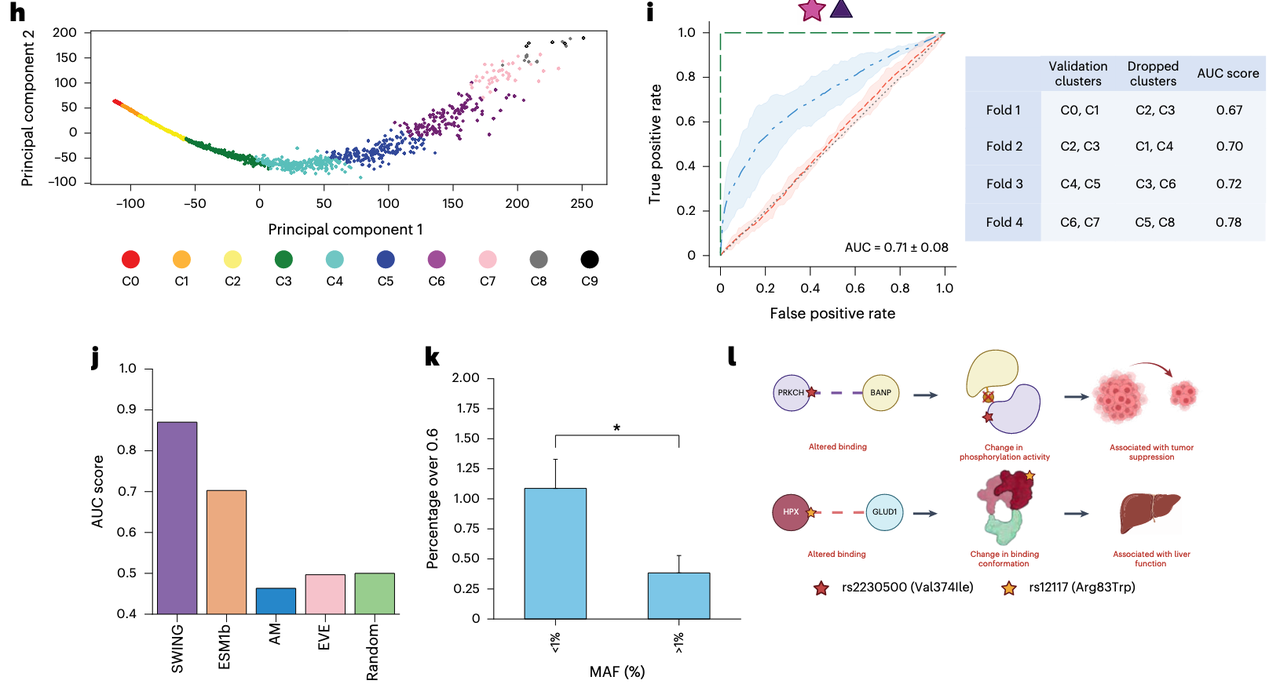
相比之下,AM、EVE、ESM1b 在相同任务中表现显著逊色(Fig. 5j)。
5.4 生物学解释与案例分析
作者展示了两个 SWING 预测的互作破坏变异实例(Fig. 5l):
PRKCH–BANP:
变异 rs2230500(Val374Ile)位于 PRKCH ATP 结合位点,同时也是 BANP 结合界面的一部分。
该变异已知与类风湿关节炎及脑小血管病相关,可能通过改变自磷酸化水平及与 TP53 的间接互作,影响细胞增殖与肿瘤抑制通路。
HPX–GLUD1:
变异 rs12117(Arg83Trp)位于 HPX 远离结合界面的区域,但 AlphaFold3 建模显示其导致构象变化,进而影响与 GLUD1 的结合。
AM 将其预测为良性,而 SWING 能识别出该种“远距构象效应”。
这一部分的意义在于,SWING 从免疫学的 pMHC 特殊互作场景,扩展到了全局蛋白互作网络的变异致病机制预测。它不仅能识别直接界面破坏,还能捕捉到通过构象变化导致的远程效应。
6. 与其他建模方法的全面对比
为了验证 SWING 框架在不同任务和泛化场景下的优势,作者将其与多种主流互作建模方法进行了系统对比(Fig. 6a–i)。
6.1 对比模型类型(Fig. 6a)
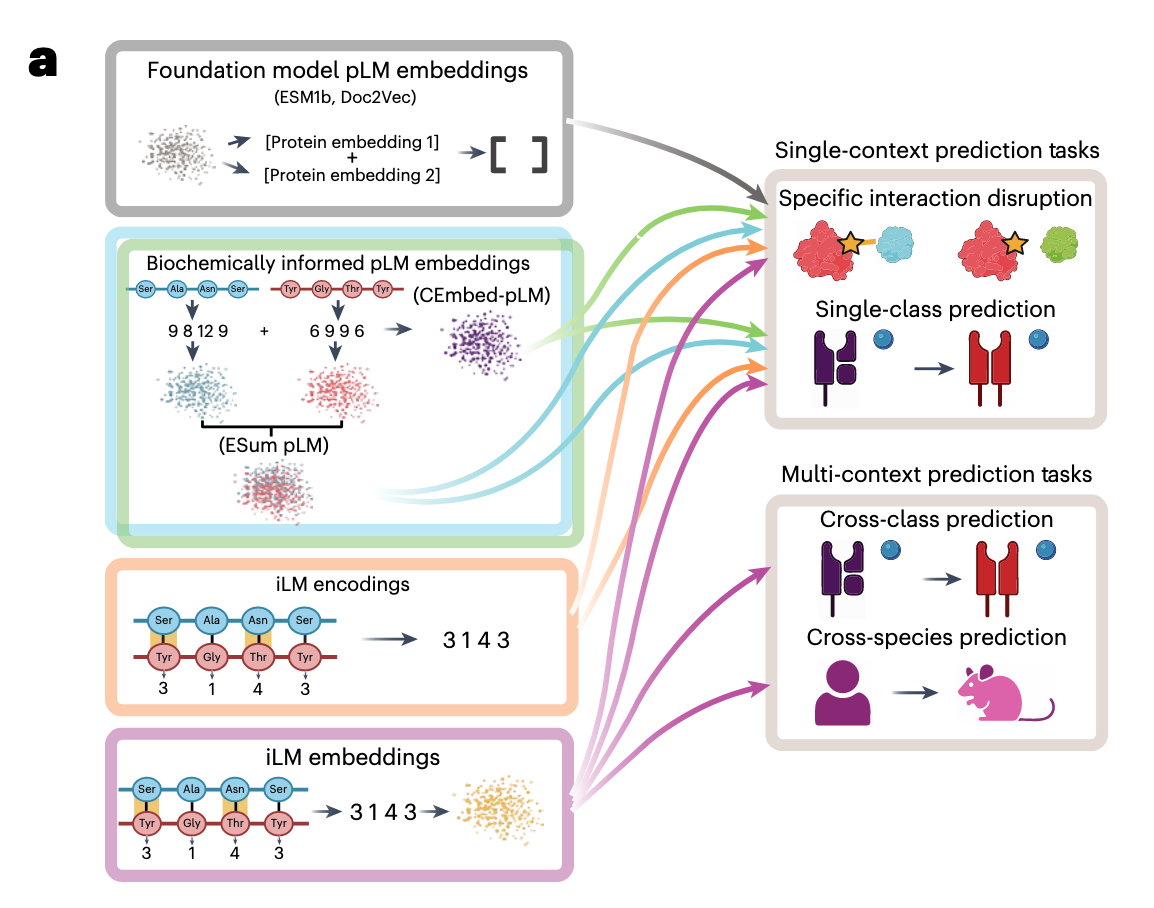
基于 pLM 的方法:
ESM1b-embedding(直接取预训练 pLM 的蛋白嵌入)
CEmbed-pLM / ESum-pLM(将配对序列的嵌入进行拼接或求和)
交互语言模型(iLM)变体:
CNN-iLM:用卷积神经网络直接学习交互语言
SWING-iLM:本文提出的方法,基于 Doc2Vec 对交互语言建模
6.2 在互作破坏预测任务中的表现(Fig. 6b)
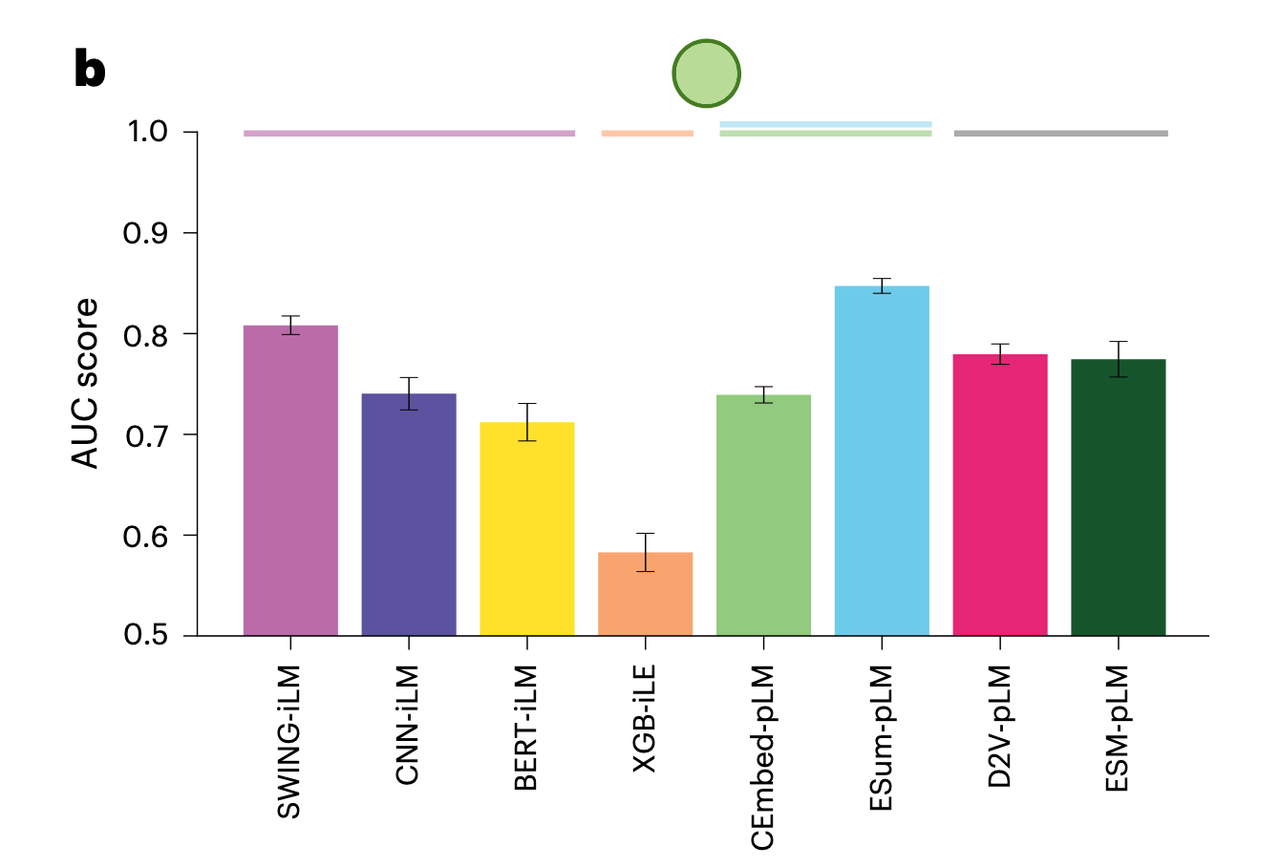
在“混合变异数据集(Mendelian + population)”的标准交叉验证(SCV)设置中:
SWING-iLM AUC ≈ 0.81,优于 CNN-iLM(~0.77)和所有 pLM 基础方法(多数低于 0.75)
随机分类器基线仅 0.5
这表明 SWING 对互作特异性模式的捕捉能力更强,而不是依赖单个序列特征。
6.3 在 MHC 结合预测中的跨等位基因泛化(Fig. 6c–i)
在多个 cross-prediction 场景下,SWING-iLM consistently 领先:
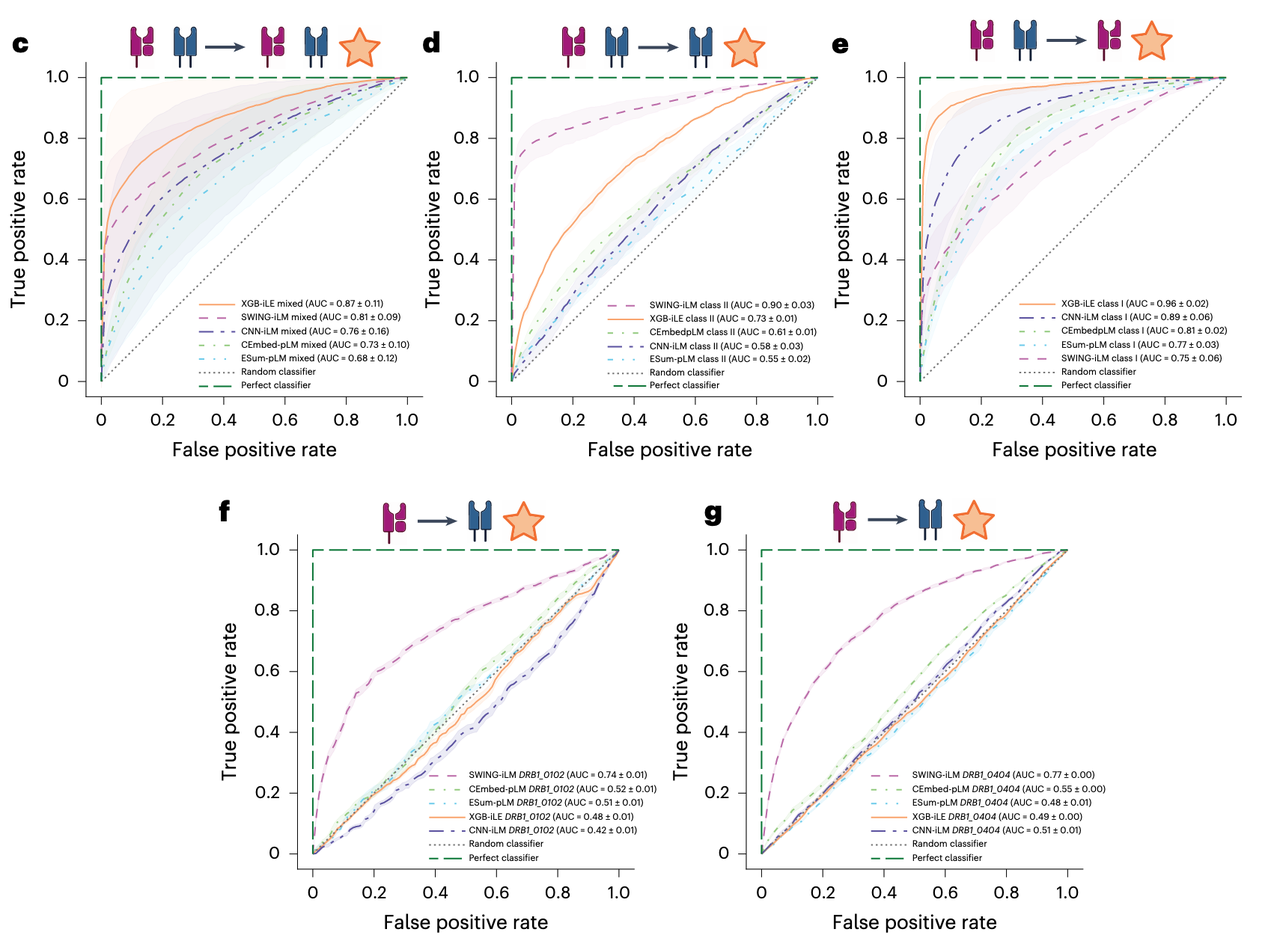
混合等位基因模型 → 未见 Class II 等位基因(Fig. 6d):SWING ~0.93,高于 CNN-iLM (~0.90) 和 pLM 方法(~0.85)
混合等位基因模型 → 未见 Class I 等位基因(Fig. 6e):SWING ~0.82,对比 CNN-iLM (~0.79)
Class I 模型 → 未见 Class II 等位基因 DRB1_0102 / DRB1_0404(Fig. 6f–g):SWING 分别保持 ~0.74 和 ~0.77
混合等位基因模型 → 小鼠等位基因 H-2-IEk / H-2-IAg7(Fig. 6h–i):SWING 分别达 0.84 和 0.91,明显优于其他方法(大部分低于 0.8)
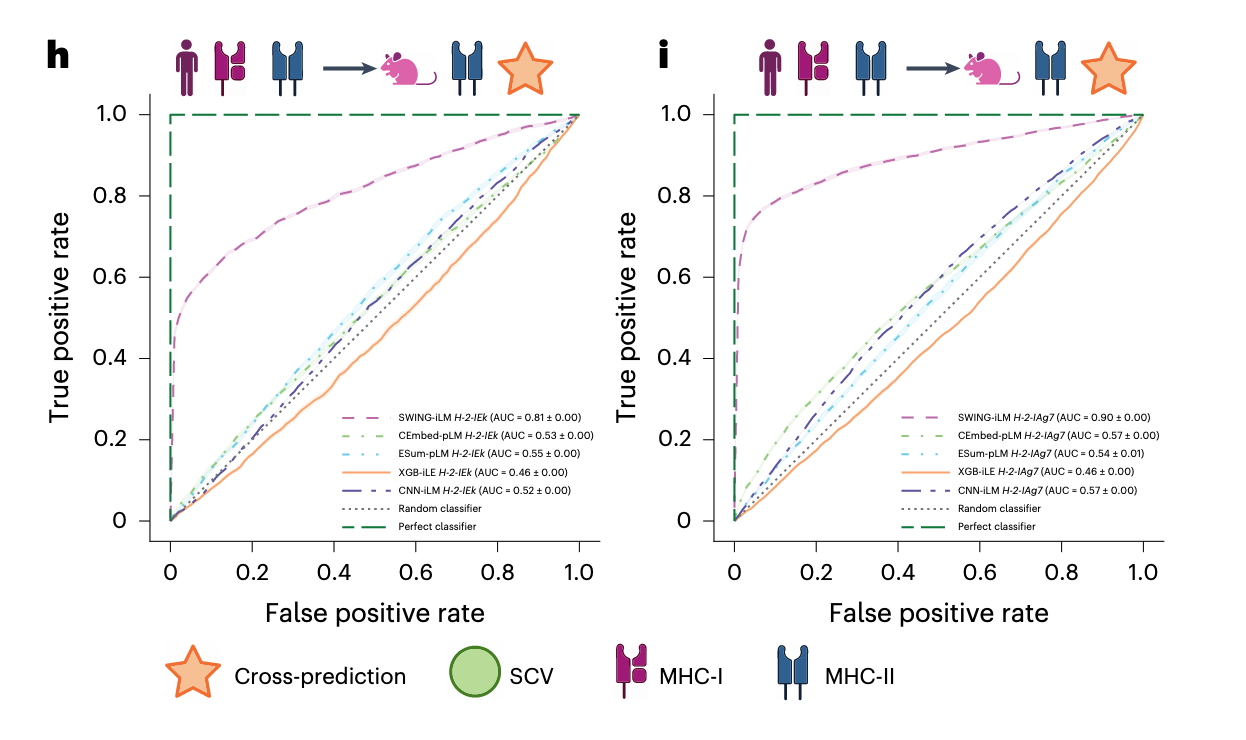
6.4 核心优势总结
生化差异驱动:编码直接来自氨基酸理化性质差异,不依赖特定结构或 motif 数据。
上下文无关性:可在跨类别、跨物种的零样本场景中保持高性能。
任务泛化性:同一嵌入可复用在免疫预测、PPI 破坏预测等完全不同的任务上。
长度无关性:不受输入序列长度限制,适应各种互作尺度。
这部分的对比结果强调了一个结论:SWING 不是在单一任务上“调优”出来的,而是一个可泛化到多类互作任务的统一框架。
7. 总结与前景
SWING(Sliding Window Interaction Grammar)通过一种全新的“交互语言”视角,将蛋白–蛋白及蛋白–肽段互作建模提升到新的高度。它的核心创新不在于更深的神经网络,而在于如何在建模前就把互作信息转化为语言结构:
方法论创新:滑动窗口提取互作片段 → 生化差异编码 → k-mer 词汇化 → Doc2Vec 嵌入 → 下游预测
性能表现:在 pMHC 结合预测中,SWING 能跨等位基因、跨类别(Class I ↔ Class II)、跨物种(人 ↔ 小鼠)实现零样本泛化
任务拓展:不仅限于免疫学,在预测氨基酸变异导致的特定互作破坏上,SWING 也优于现有主流 VEP 工具
对比优势:在多种 cross-prediction 场景下,SWING consistently 高于 CNN-iLM 及基于 pLM 的嵌入方法
未来潜力:
免疫学应用:可辅助疫苗设计、稀有 MHC 等位基因免疫应答预测,以及疾病相关免疫表位的快速筛选
药物研发:用于小分子–蛋白、抗体–抗原等互作的亲和力预测,缩短早期筛选周期
结构未知互作推断:在缺乏高分辨率结构数据的情况下,利用序列级互作语言实现快速推断
大规模互作网络解析:在系统生物学层面,帮助识别关键互作节点和致病变异靶点
一句话总结:
SWING 不是一个“针对某个问题的专用模型”,而是一个能跨任务、跨类别、跨物种的通用互作语言框架。它为蛋白质设计与功能预测提供了新的思路——先理解“对话”,再设计“角色”。
原文链接:https://doi.org/10.1038/s41592-025-02723-1
延伸阅读
本文属于 AI4S文献 栏目。
返回 AI4S文献 → 去公众号阅读完整版 →