ICML 2025 Spotlight|力文所 Pallatom 开启全原子蛋白设计新范式
在前不久结束的 ICML 2025(国际机器学习大会)上,有一篇来自中国初创企业 力文所(Levinthal) 和复旦大学的合作论文引起了广泛关注。在这个以传统机器学习为主的顶会上,这篇做蛋白质设计的论文不仅入选,还被评为 Spotlight 论文。今天就和大家分享一下这个值得关注的工作:**《**P(all-atom) Is Unlocking New Path For Protein Design》。


为什么这篇文章值得我们细读?
因为它提出的 Pallatom 模型,开辟了一条全新的路径:直接在全原子层面(all-atom)进行蛋白质生成。
这意味着我们不再停留在“预测骨架”或“序列-结构分离”的阶段,而是真正走向 原子级精度的蛋白设计。
对于蛋白质设计这个快速发展的前沿领域而言,这是一件非常漂亮、也非常有意义的工作。在英伟达最新的技术博客中,在蛋白质基础模型中,Pallatom 被放在了与这些巨头模型并列的位置,也一方面证明了Pallatom的价值。

🔗Palatom原文链接:https://doi.org/10.1101/2024.08.16.608235
第一部分:背景介绍
在蛋白质设计领域,有一个核心问题:
怎样同时得到“序列”和“结构”?
传统上,科学家们常走两条路:
从序列出发:预测出三维结构(P(structure | seq)),代表成果是 AlphaFold 系列。
从骨架出发:在给定 backbone 上寻找能折叠成该结构的序列(P(seq | backbone)),代表工具是 ProteinMPNN 等。
这两种思路极大推动了蛋白设计的发展,但它们都有一个共同的缺陷:都没有真正把“全原子(all-atom)层面的信息”建模进来。
在蛋白质里,侧链的朝向、氢键的网络、疏水/亲水分布,这些都决定了最终的稳定性和功能。如果只停留在 backbone 层面,或者单纯关注序列与骨架匹配,很多精细的物理化学特征会被忽略。
第二部分:Pallatom 的核心理念
Pallatom 的核心创新在于:直接学习 P(all-atom) ——也就是说,模型直接在全原子坐标空间中建模,把结构与序列的关系一次性捕捉。
这和以往方法的区别在哪里?
过去的方法往往是“拆开建模”:先生成 backbone,再去补序列;或者先从序列出发,再通过语言模型或能量函数去优化。
Pallatom 的观点是:蛋白质的全原子坐标本身,已经同时包含了序列和结构信息。只要我们能在原子层面正确建模,就不需要再额外拼接其他模块。
📌 配图对应(Figure 1A):这里展示了 Pallatom 的整体架构。它采用了一个 dual-track 框架:

Residue-level track:在残基层面建模,关注全局信息。
Atom-level track:在原子层面建模,关注局部几何与化学细节。
这两条轨道并不是平行工作的,而是通过 AtomDecoder 单元不断交互:
残基级别的表示会“广播”到原子层面;
原子级别的特征再被“回收”,反馈到残基层面;
经过多层迭代,结构和序列的特征逐步融合。
这种“往返式”更新的思路,保证了 backbone 和 sidechain 都能被精细建模,避免了传统方法里“结构与序列脱节”的问题。
Pallatom 不再把蛋白质拆分成骨架和序列两个独立问题,而是通过全原子建模,把两者真正地绑在了一起。
第三部分:关键技术亮点
在 Pallatom 的框架下,有三个关键技术点值得特别强调。
1. atom14 表示法:统一全原子表示
在蛋白质生成任务中,一个最大难题是:在序列未知之前,你根本不知道系统到底有多少原子。
不同的氨基酸侧链差别很大,比如丙氨酸 (Ala) 只有一个甲基侧链,而色氨酸 (Trp) 的侧链则庞大复杂。
Pallatom 提出了一个巧妙的解决方案:atom14 表示法。
它为每个残基固定分配 14 个“虚拟原子位置”,无论真实氨基酸是什么。
当序列确定后,才会丢弃多余的虚拟原子,只保留真实的化学结构。
这样一来,模型在一开始就能用统一的点云来学习,避免了“原子数随序列变化”的麻烦。

2. Dual-track 框架与 AtomDecoder:往返交互的秘密武器
Pallatom 的 backbone 在 残基层面和原子层面同时建模,但真正的关键是 AtomDecoder 单元。
在解码过程中,残基层面的特征会“广播”(broadcast)到原子层面;
原子层面的几何特征再被“回收”(recycle),反馈给残基层面;
通过多次迭代,这种往返交互不断细化了坐标预测。
这种机制保证了 backbone 和 sidechain 的耦合建模,避免了传统方法里“骨架正确但侧链乱”的问题。

3. SeqHead 模块:从几何到序列的桥梁
即使有了 atom14 表示法,模型生成的还是一团“坐标云”,要把它转化为生物学意义上的氨基酸序列,还需要一个关键步骤。
这就是 SeqHead 模块:
它从每个残基的原子嵌入中提取信息;
通过一个分类层预测 20 种氨基酸的分布;
最终实现“几何 → 序列”的映射,让模型既能生成结构,也能生成对应的序列。
一句话总结:
atom14 解决了输入表示的问题,AtomDecoder 解决了结构建模的问题,SeqHead 解决了序列输出的问题。三者结合,让 Pallatom 真正做到了全原子、端到端的蛋白质生成。
第四部分:实验结果与性能对比
一个模型再有创意,最终还是要看能不能“打得过”现有方法。研究团队对 Pallatom 做了系统评估,结果相当亮眼。
1. 设计性(Designability)显著提升
Pallatom 在 CO-DESIGN 模式下的设计成功率(DES-aa)高达 85%+,远超 Protpardelle(30%)、ProteinGenerator(43%)、Multiflow(62%)。
这说明 Pallatom 生成的序列与全原子结构是高度一致的,不需要额外的“修修补补”。
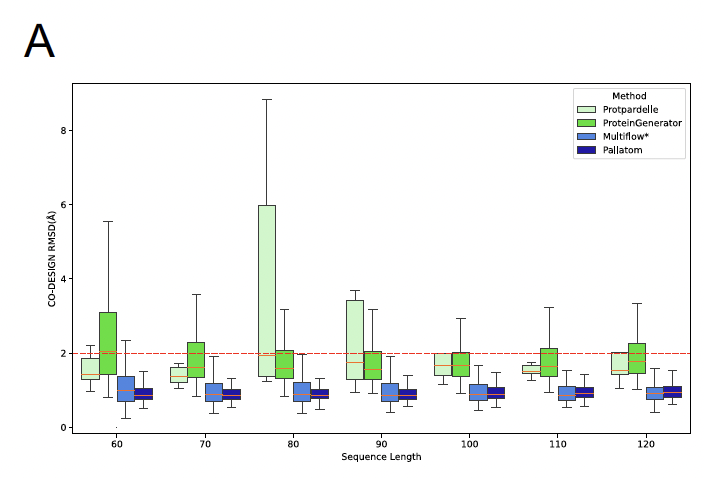
2. 多样性(Diversity)全面领先
Pallatom 在结构和序列的多样性上,都远远超过其他方法。
具体来说,生成的结构数量更多、分布更均匀,序列也展现了更广的组合空间。

3. 新颖性(Novelty)更高
Pallatom 生成的结构与天然蛋白(PDB 中的已知结构)的相似度更低(NOV-str 较小)。
这意味着它不仅能“复刻”自然界的蛋白,还能探索新的 fold 空间。

4. 对比结果一览
在 Table 1 的对比中,Pallatom 在三大维度(设计性、多样性、新颖性)几乎全面领先,甚至超越了专门为 backbone 设计优化的 RFdiffusion。

一句话总结:
Pallatom 不是“只在某一维度强”,而是做到了全方位平衡——既能设计成功率高,又能保证多样性,还能探索真正新颖的结构空间。
第五部分:消融实验——为什么 Pallatom 有效?
研究团队专门做了消融实验,逐一去掉 Pallatom 的核心组件,来看看性能会发生什么变化。结果很有意思。
1. atom14 表示法的必要性
如果把 atom14 替换成传统的 hybrid14(即对 20 种可能的侧链坐标做叠加),设计性(DES-aa)从 87% 降到 5%!
说明 hybrid14 的“序列驱动”思路在扩散建模中并不适用,反而导致序列和结构严重脱节。
结论:atom14 是 Pallatom 的根基,它保证了生成过程中序列与结构能自然耦合。
2. 回收机制(Recycling)的重要性
- 如果去掉 recycling 机制(woRC),backbone 的指标变化不大,但全原子设计性(DES-aa)从 87% 掉到 21%。
说明 recycling 对 backbone 影响有限,但对 序列生成至关重要。它让残基层面和原子层面的信息能够往返流动,保证生成的序列和几何结构相互匹配。
3. 结论:架构设计的必要性
这些消融结果表明,Pallatom 的成功不是靠“大模型堆料”获得的,而是 核心设计点(atom14 + recycling)共同作用的结果。
atom14 解决了“如何统一表示”的难题;
recycling 确保了“序列与结构的耦合”。

Pallatom 的架构设计具有高度的针对性和必要性,这也是它能全面超越现有方法的根本原因。
第六部分:影响与展望
Pallatom 并不仅仅是一个“模型性能更好”的工作,它更重要的是:开启了全原子层面蛋白质生成的新范式。
1. 对科学研究的意义
过去的 backbone 级方法虽然能设计出稳定骨架,但对侧链的处理往往粗糙。
Pallatom 直接在全原子层面工作,这意味着它能捕捉到 氢键网络、疏水/亲水分布、电荷相互作用 等精细物理化学特征。
这让生成的蛋白不仅在几何上合理,更在能量学和功能上具备真实性。
2. 对应用的价值
酶设计:催化位点的活性依赖于原子级别的精确定位,Pallatom 的 all-atom 能力正好契合。
药物结合口袋设计:小分子结合需要原子尺度的空间匹配和电荷补偿,全原子生成模型可以直接生成带有功能性 pocket 的蛋白。
复杂体系设计:例如蛋白-蛋白相互作用界面、纳米材料构建,Pallatom 的思路都能拓展。
3. 未来方向
论文最后也提到,未来 Pallatom 的潜力在于:
扩展到更大规模、更复杂的体系,比如多聚体或膜蛋白;
改进架构,让模型更加通用和高效;
结合实验验证,推动生成结果走向真正的 wet-lab 应用。
Pallatom 不只是一个模型,而是一种全新的蛋白设计范式,它让我们离“原子级别的精准蛋白工程”更近了一步。
蓝极点评:
在蛋白质设计这条路上,我们已经走过了从 序列到结构、从 骨架到序列 的阶段。
但只有当我们进入 全原子层面(all-atom),蛋白质的真实物理和功能才真正浮现。
Pallatom 的意义,就在于它首次证明:
通过直接建模 P(all-atom),可以一步到位生成既有结构又有序列的高质量蛋白。
它不仅在指标上全面超越现有方法,更重要的是开辟了新的研究方向——让蛋白设计从“近似”走向“原子级精度”。
无论是酶工程、药物设计,还是全新的蛋白折叠空间探索,Pallatom 都展示了巨大的潜力。
或许不久的将来,我们会看到更多这样的 all-atom 模型,真正把“设计蛋白质”变成一门精细可控的工程学科。
延伸阅读
本文属于 AI4S文献 栏目。
返回 AI4S文献 → 去公众号阅读完整版 →