从静态到动态:AI 在蛋白质动力学建模中的新进展
在过去几年,AlphaFold2 的横空出世几乎改变了整个结构生物学的研究格局。一个氨基酸序列,几乎可以“一键”得到对应的三维结构——这在十年前是难以想象的。
但是,结构预测的革命同时也暴露了一个新的问题:蛋白质并不是一个固定不动的雕塑,而是一个在能量地形上不断“呼吸”、在多种构象之间切换的动态分子机器。静态结构虽然能告诉我们“长什么样”,却往往难以揭示“怎么动、为什么动”。而这些动态,恰恰是催化、结合、信号转导等功能的关键所在。

今天要介绍的这篇综述文章(《Generation of protein dynamics by machine learning》)正是聚焦于这个前沿问题:如何用机器学习的方法,不仅预测蛋白质的单一构象,而是预测其动态构象集合(conformational ensembles)。文章中的配图(Figure 1)非常直观地把研究方向总结为四条主线:
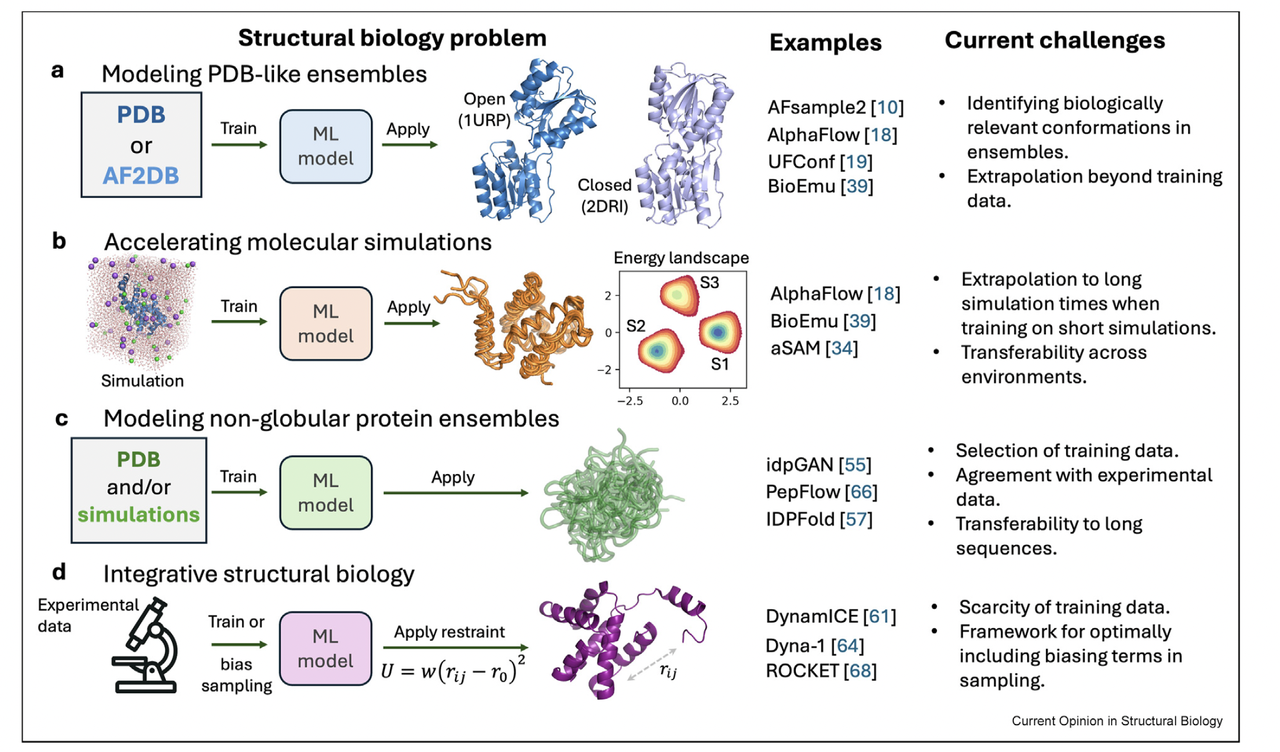
从单一结构到多态结构(PDB-like ensembles);
加速分子动力学模拟(accelerating MD);
非球状蛋白和无序区域的建模(non-globular proteins);
融合实验约束的综合模型(integrating experimental data)。
接下来,我们就结合这张图,逐一解读这四个方向的研究亮点与挑战。
1. PDB-like Ensembles:从“一个结构”到“多个状态”
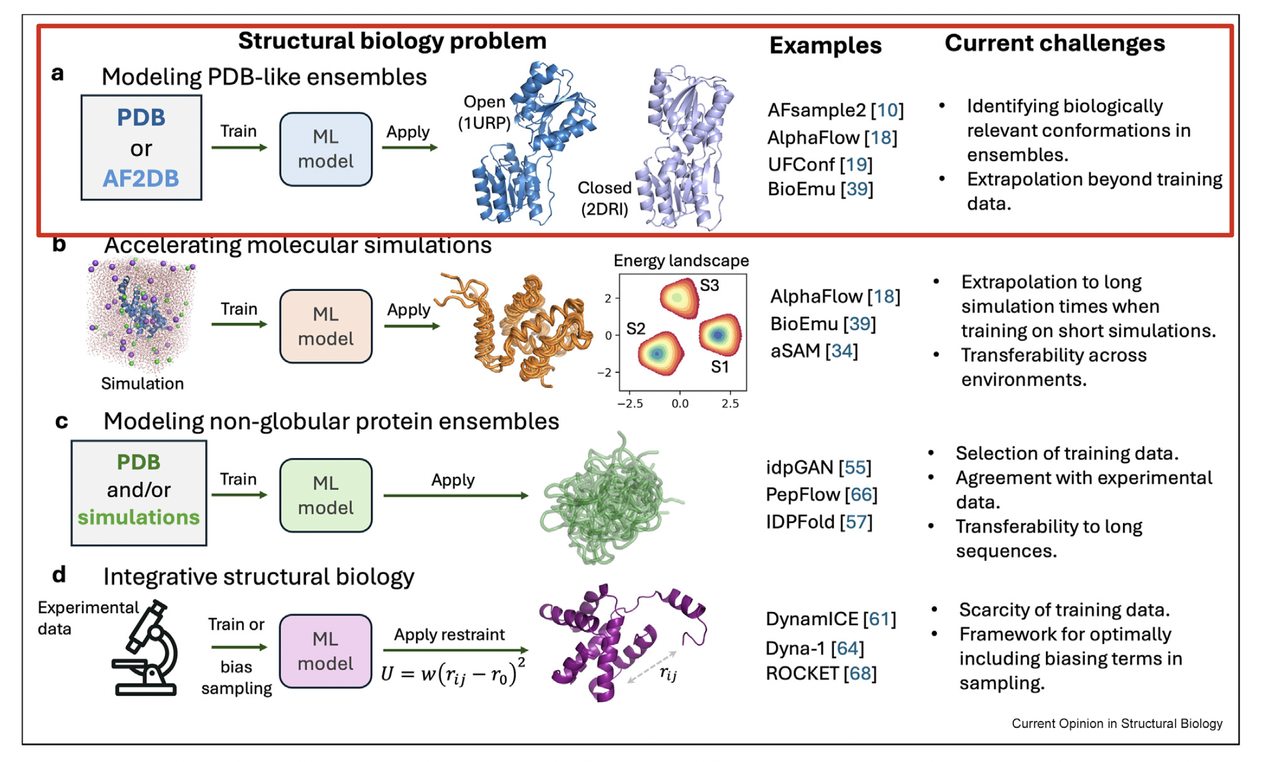
看 Figure 1(a),我们可以直观感受到作者想传递的核心信息:同一个蛋白质,并不是只有一个固定结构,而是可能存在多种功能相关的状态。在 PDB 数据库里,其实早就能找到一些蛋白的多态结构——比如同一个蛋白在有配体(holo)和无配体(apo)条件下的不同状态,又或者激活/非激活状态的跨膜受体。
然而,AlphaFold2 的局限在这里就显现出来了。它的训练目标是 “一条序列 → 一个最可能的结构”,因此它很难自然地给出“一个序列 → 一组结构分布”。这也是为什么它在预测 holo/apo 或 fold-switching 蛋白时表现不佳。
研究者们于是开始尝试各种“曲线救国”的办法:
MSA 修改:通过改变输入的多序列比对(MSA),可以诱导 AlphaFold 给出不同的构象。比如 AFsample2 会随机屏蔽 MSA 列,从而在开放/闭合状态的蛋白里得到多态结构。
聚类策略:像 AF-cluster,通过对 MSA 做聚类,捕捉到大构象转换(如 KaiB 这种可折叠切换的蛋白)。
生成模型(Generative Models):这是更“根本”的方向,把 AlphaFold 从确定性回归模型转化为生成式模型。例如 AlphaFlow、UFConf 这样的 diffusion model,可以直接学到“同一序列对应多个可能构象”的分布。
这些方法的亮点在于,它们开始让计算机模型能够“想象”蛋白质的不同功能状态,而不是只锁定在唯一的静态解。但它们的瓶颈也很明显:
目前还难以保证不同状态的相对概率是否正确,也就是说,哪个构象更常见、哪个只是低概率事件,模型给不出准确答案。
对超出训练集的折叠切换蛋白,泛化性依旧不足。
总结来说,Panel (a) 展示的是整个领域的第一步跨越:从单一结构预测,走向构象集合生成。这一步虽然还不完美,但它让我们看到了用 AI 捕捉蛋白质“多态性”的可能。
2. Accelerating Molecular Simulations:让分子动力学“加速跑”
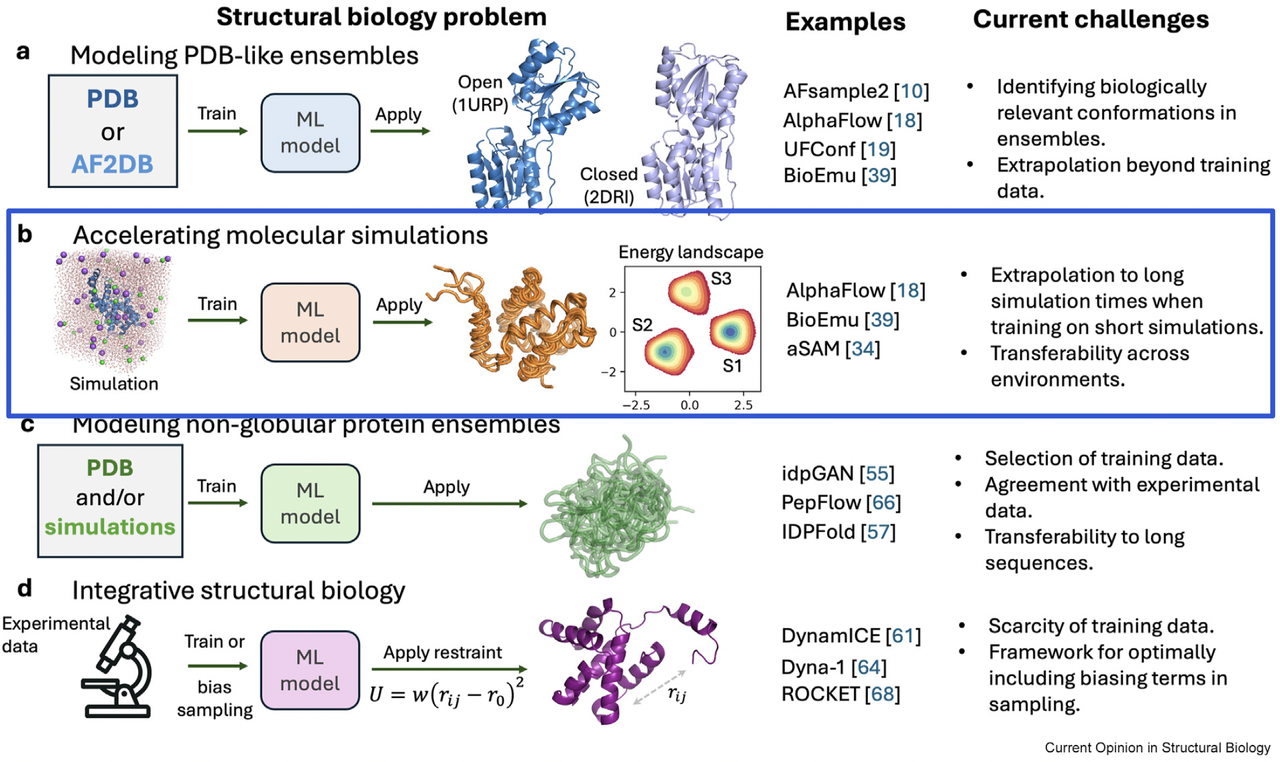
如果说 Panel (a) 的主题是“如何生成更多状态”,那么 Panel (b) 就回答了另一个问题:如何更快地采样这些状态?
看 Figure 1(b),图示了能量地形(energy landscape),上面分布着不同的构象状态。传统上,我们依赖分子动力学模拟(MD)来探索这个能量地形,让蛋白质在时间轴上不断演化,逐步采样到不同状态。问题是,MD 计算量极其庞大,尤其是想要捕捉到大尺度的构象变化时,可能需要耗费几个月甚至更久的超级计算资源。
机器学习提供了几条加速思路:
- 基于 MD 数据训练生成模型
这些模型直接从 MD 数据中学习到统计分布,然后能快速生成彼此独立的构象样本。这样就像“跳过”了冗长的时间积分,直接拿到模拟结果。
代表:AlphaFlow 在 ATLAS 数据集上训练,能高效生成不同蛋白的结构集合。
神经网络粗粒化力场(CG force fields)
通过神经网络拟合原子间作用力,把全原子的复杂系统压缩成简化模型,再进行模拟,既快又保留主要物理特征。
- “时间跳跃”模型
与其一步步积分时间,不如让生成模型学会直接预测“未来更远时间的状态”。这样就能跨越原本需要成千上万步积分才能到达的时间尺度。
- 混合增强采样
比如 AlphaFold2-RAVE,把 AF2 生成的多态结构输入到 MD,再用机器学习方法学习反应坐标,帮助 MD 更高效地在构象空间里采样。
这些方法有各自的亮点与局限:
生成模型采样快,但通常只能给出热力学分布(各状态出现的概率),却不一定能还原动力学过程(不同状态之间的转换速率)。
时间跳跃模型能提供动力学信息,但对复杂能量地形(多稳态系统)还不够稳健。
混合方法能恢复多个状态,但普适性和可迁移性还不足,每个新体系都要重新训练。
总结来看,Panel (b) 告诉我们:机器学习不是要取代分子动力学,而是让它“加速跑”。通过生成模型和混合策略,我们或许能在保证物理真实性的同时,把过去几个月的模拟压缩到几小时甚至更短。
3. Non-globular Proteins:让“无序”也有可预测性
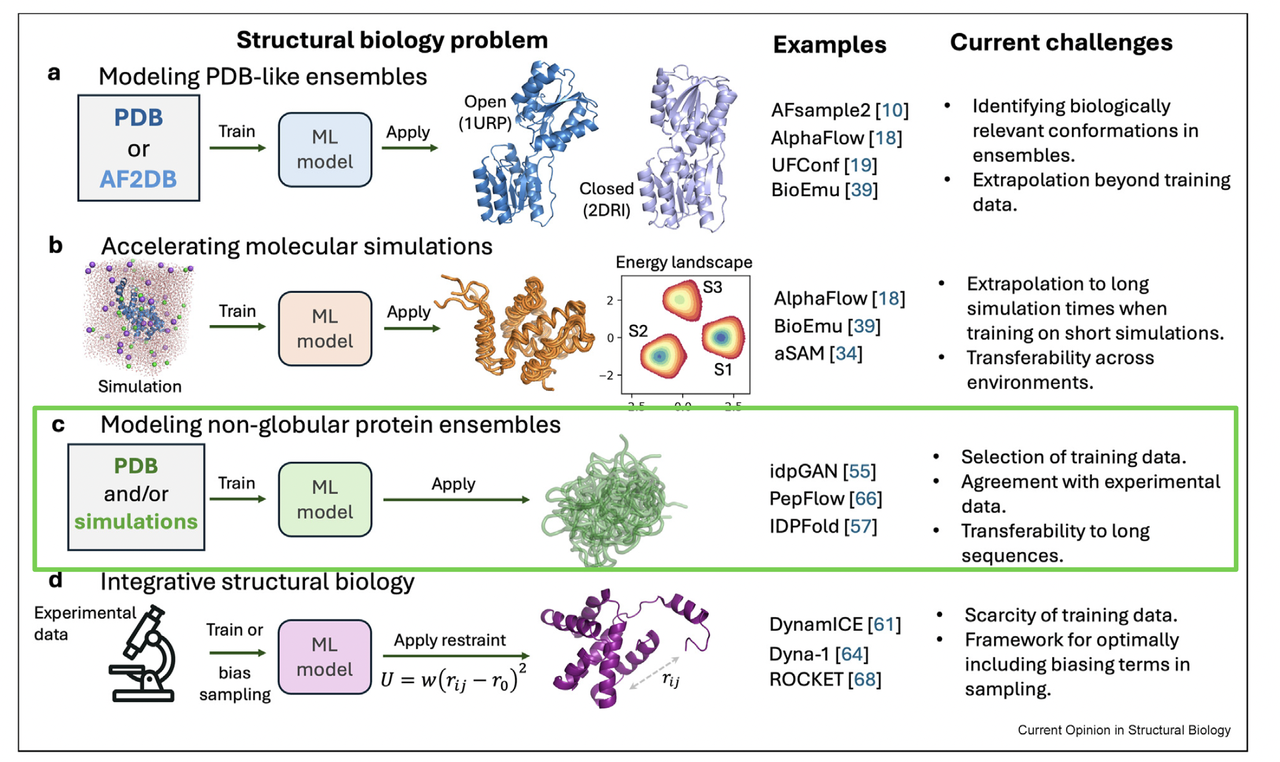
在前面,我们主要讨论了类似“球状”蛋白(globular proteins)在不同状态下的多态性。但在生物体内,还有一类蛋白更加棘手:非球状、甚至完全无序的蛋白。
看 Figure 1(c),你会看到那些长链状、松散的构象,这就是 IDRs(intrinsically disordered regions) 的典型特征。它们不像球状蛋白那样有稳定折叠的三维结构,却在调控、信号传导、相分离等过程中发挥关键作用。问题是:
实验测不到清晰结构,因为它们本身就是一堆不同构象的混合。
AF2 给不出合理预测,虽然能识别出哪些片段是无序的(通过低置信度分数),但预测出来的构象往往过于“理想化”,和真实的动态集合相差甚远。
于是,研究者们开始尝试开发专门的 ML 方法来应对这类系统:
idpGAN:用 coarse-grained 模拟数据训练生成模型,可以快速生成不同 IDR 的结构集合。
IDPFold:采用“混合训练策略”,先用 PDB 和 NMR 的高分辨率数据预训练,再用低分辨率模拟数据微调,从而能捕捉到 IDR 的实验可观测特征(比如化学位移、半径分布)。
BioEmu:虽然并没有针对 IDR 专门训练,但在测试时展现了“零样本”能力,能生成接近实验的构象集合。这说明“混合数据+生成模型”是一条很有前景的路线。
这些工作揭示了一个重要趋势:未来的蛋白质建模,不仅仅是给出“一个漂亮的结构”,而是要能生成“一个构象集合”,并且让集合的统计特征尽量吻合实验观测。
挑战也很现实:
数据稀缺:无序蛋白在 PDB 里几乎没什么代表性结构。
泛化难题:如何处理超长 IDR(>200 氨基酸),目前几乎只能依赖粗粒化模拟,精细度受限。
总结来说,Panel (c) 向我们展示了一个新前沿:机器学习正在帮助我们用统计学的方式理解“无序中的秩序”。这不仅有助于解释 IDR 的生物学功能,也可能为神经退行性疾病(如与 amyloid 相关的病理)带来新的研究工具。
4. Integrating Experimental Data:用实验“校正”AI 的想象力
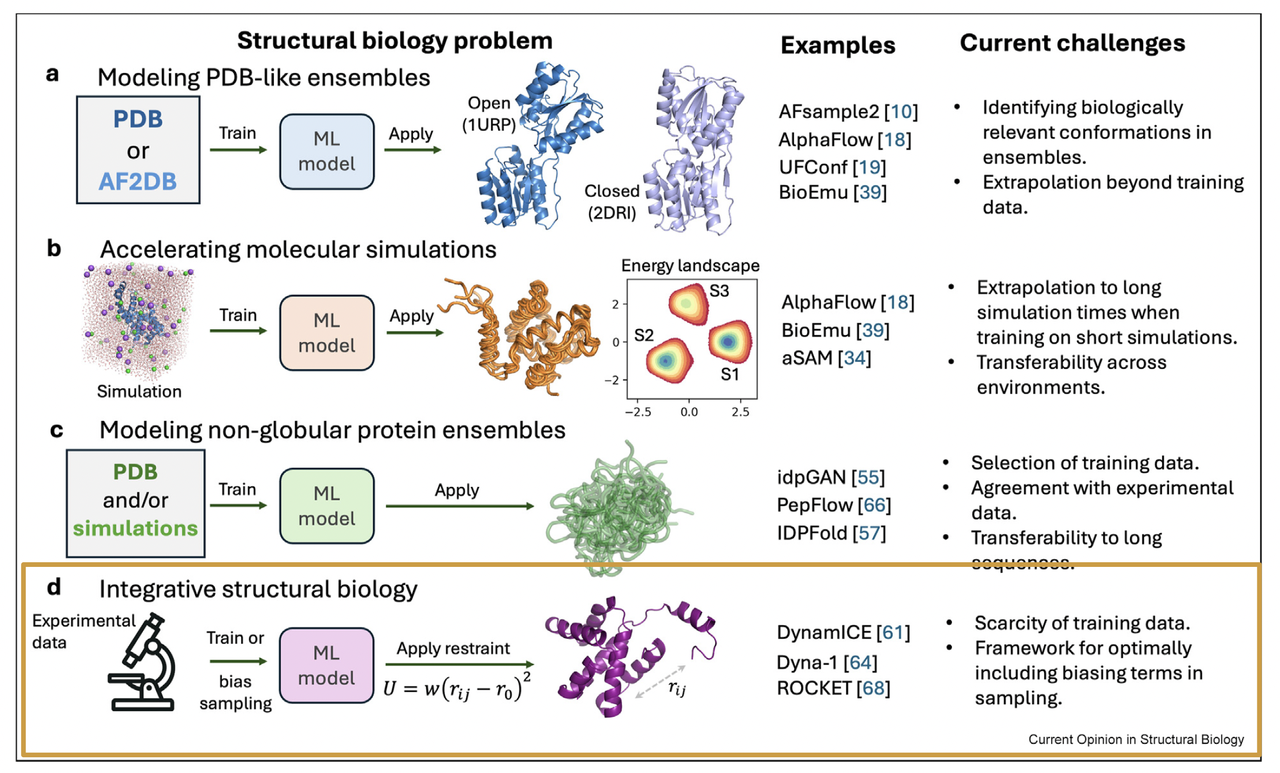
到这里,大家可能会问:机器学习模型预测出来的这些构象集合,究竟有多接近真实?这正是 Panel (d) 想回答的问题。
看 Figure 1(d),配图里展示了实验数据(比如 NMR、Cryo-EM、红外光谱等)与机器学习模型结合的过程。思路很简单:
单靠 AI,它可能生成一些“看起来合理”的构象,但未必反映真实概率分布。
单靠实验,又往往捕捉不到完整的动力学集合,只能提供一些“投影信息”。
两者结合,才能让预测既高效,又更贴近真实世界。
目前主要有两条策略:
训练阶段引入实验约束
DynamICE:在训练时直接用 NMR 数据对模型进行微调,让生成的构象集合符合实验观测。
DEERFold:在 AlphaFold 基础上引入双电子自旋共振(DEER)数据,得到更符合实验的结构。
Dyna-1:用大规模 NMR 弛豫数据训练,直接捕捉毫秒级的蛋白动力学。
推理阶段引入实验引导
不改变模型本身,而是在生成时给它“加偏置”。
例如 idpSAM 在采样过程中引入 radius-of-gyration 约束,IDPForge 更进一步,能结合 NMR 数据、Rg 分布、甚至已有的折叠域信息。
在球状蛋白里,也出现了类似思路:比如 ROCKET 算法,让 AlphaFold 生成的模型更好地贴合 X-ray 或 Cryo-EM 数据;实验引导的 AlphaFold3 则把电子密度直接融入扩散过程,使动态区域更接近实验结果。
这两种策略各有优势:
训练时引入约束,能让模型从根本上“学会”物理规律,但需要大量高质量实验数据。
推理时加入偏置,灵活性更高,也能针对不同实验条件快速适配。
总结来说,Panel (d) 向我们展示了一个未来愿景:机器学习与实验不再是平行的两条路,而是互相补充的伙伴。AI 提供速度与想象力,实验提供真实性与校正能力,两者结合有望让蛋白质动力学建模迈向真正的“精准预测”。
总结:从静态雕塑到动态生命
回顾全文,我们可以看到一个清晰的趋势:
AlphaFold2 解决了“蛋白质长什么样”的问题;
而新一代的机器学习方法,正在解决“蛋白质是怎么动的”这一更关键的问题。
无论是 Panel (a) 的多态生成,Panel (b) 的模拟加速,Panel (c) 的无序蛋白建模,还是 Panel (d) 的实验结合,它们都指向同一个未来:
AI 将帮助我们不仅看见蛋白质的“照片”,还将捕捉到它们的“电影”。
对于蛋白质设计和药物研发来说,这意味着我们将更有可能预测并调控蛋白质的功能状态,为靶向设计和精确干预开辟全新道路。
🔗原文链接:https://doi.org/10.1016/j.sbi.2025.103115
延伸阅读
本文属于 AI4S文献 栏目。
返回 AI4S文献 → 去公众号阅读完整版 →