EvoBind2:基于蛋白质序列信息设计不同长度的线性与环状肽结合物

作者:Qiuzhen Li, Efstathios Nikolaos Vlachos, Patrick Bryant
单位:斯德哥尔摩大学 Wenner-Gren研究所 & Science for Life Laboratory
预印本链接:https://doi.org/10.1101/2024.06.20.599739
一、研究背景
蛋白质的结构预测已因AlphaFold等AI技术取得突破性进展。然而,如何从序列信息直接设计具有功能(如结合能力)的蛋白质或肽分子,仍然是合成生物学和蛋白质工程中的重大挑战。
传统的结合物设计依赖于:
靶标蛋白的三维结构
已知的结合位点
事先定义的肽长度或骨架模板
这种依赖限制了新靶点的开发和筛选能力。为了解决这个问题,本文提出了一个“盲设计”策略(EvoBind2),即在没有结构或靶点位置信息的情况下,直接从序列设计肽类结合分子。
二、与传统方法的对比
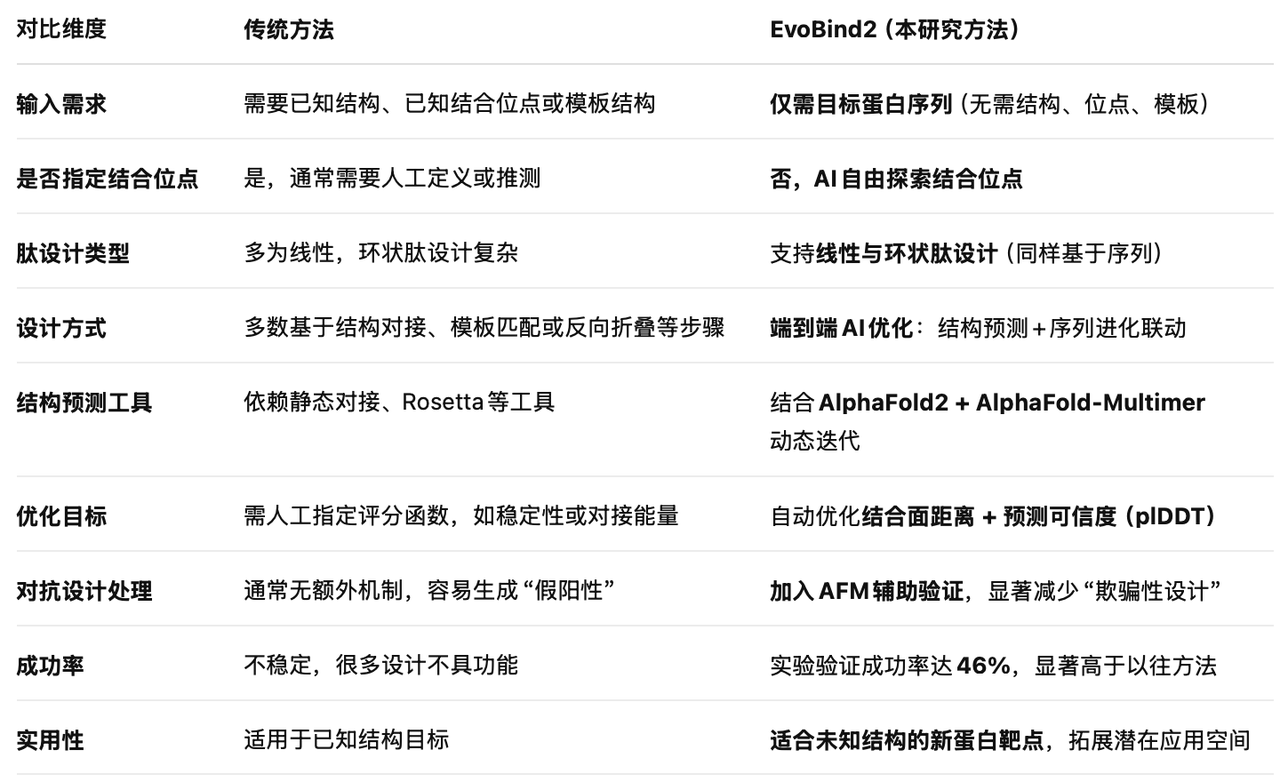
🔍 核心创新点
- 全盲设计(Blind Design)能力:
EvoBind2是目前首个实现无需结构、位点、模板信息、即可全自动生成肽结合物的系统。
- 结构预测+进化优化联动:
通过结构预测驱动序列迭代,结合预测可信度和几何距离进行优化,增强设计合理性。
- 对抗性过滤机制:
引入AlphaFold-Multimer辅助交叉验证,有效剔除“高评分但不真实结合”的序列。
三、“无结构输入设计”的科学难点与意义
🧩 科学难点
1. 缺乏结构信息导致搜索空间极大
在没有已知结构或结合位点指导的情况下,肽设计必须同时决定肽的位置、结构与序列,这使得设计空间呈指数级增长(20^L 种序列 + 无限种结构构象)。
对于传统方法而言,这相当于在没有地图的情况下寻找目标。
2. 无共进化信息,降低预测可靠性
AlphaFold2 等工具在结构预测上依赖于多序列比对(MSA)中的共进化信号。
设计新肽时,仅有目标蛋白序列,缺乏MSA背景,这导致结构预测准确性下降。
3. 高评分≠真实结合:对抗性设计问题
AI模型可能“过拟合”某些结构特征,生成“看起来可信”的但实际上无功能的序列(即“adversarial designs”)。
判断哪些预测结果是“假阳性”非常困难,需要额外机制避免模型欺骗。
4. 评估标准缺失
- 传统的评分标准如 plDDT、interface RMSD 等,在这种完全盲设计中难以预测真实亲和力,缺少可靠的后验验证机制。
🌟 科学意义与潜力
1. 摆脱结构依赖,解放设计范围
- 绝大多数天然蛋白无已解析结构,EvoBind2 的方法可用于任意蛋白靶点,极大拓宽设计空间,推动生物工程的“结构自由化”。
2. 实现从“功能导向”而非“结构导向”的设计
- 传统设计路径是“结构→功能”,而EvoBind2探索“序列→功能”,更贴近生物自然进化机制。
3. 推进通用蛋白药物开发平台
- EvoBind2使得无需结构就能设计潜在结合物药物成为可能,为靶点验证、疫苗设计、抗体工程等提供新工具。
4. 推动蛋白AI模型的交叉验证机制发展
- 本研究引入AlphaFold-Multimer作为“第二判断者”,为AI预测建立了对抗过滤思路,这对未来蛋白AI模型的“可信度体系”建设具有示范作用。
三、方法概述:EvoBind2 设计框架
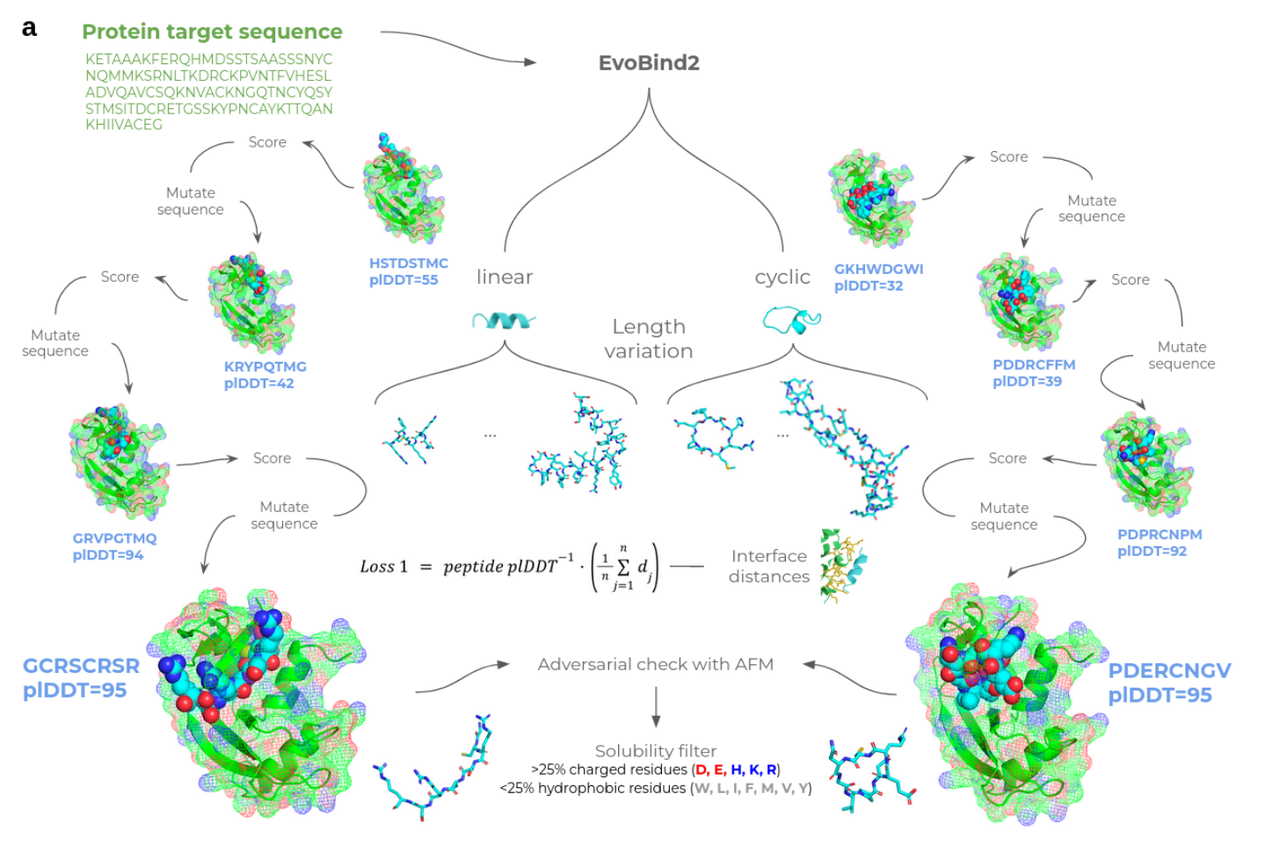
- 核心技术
EvoBind2: 一个基于AlphaFold2和AlphaFold-Multimer的AI设计平台
输入: 目标蛋白的氨基酸序列
输出: 具有预测高亲和力的线性或环状肽结合物(长度8–20残基)
优化策略
- 利用(1)结构预测可信度(plDDT)+ (2)肽与蛋白距离最小化的损失函数进行迭代优化
$$ \text{Loss 1} = \left(\text{peptide } plDDT^{-1}\right) \cdot \left( \frac{1}{n} \sum_{j=1}^{n} d_j \right)
$$
- 使用AlphaFold-Multimer辅助验证,避免生成虚假高评分但不结合的肽序列
$$\text{Loss 2} = \left( \text{peptide } plDDT^{-1} \right) \cdot \left( \frac{1}{m} \sum_{i=1}^{m} d_i + \frac{1}{n} \sum_{j=1}^{n} d_j \right) \cdot \frac{1}{2} \cdot \Delta COM$$
$$\text{peptide} \ plDDT^{-1}$$: 预测肽链的平均可信度(plDDT)的倒数,可信度越高,该项越小(更优)
$$\frac{1}{m} \sum_{i=1}^{m} d_i$$ : 设计中肽原子与靶蛋白关键位点之间的平均最短距离
$$\frac{1}{n} \sum_{j=1}^{n} d_j$$: 设计肽与整体蛋白界面的平均距离
$$\frac{1}{2}$$: 权重因子,标准化整个损失值
$$\Delta COM$$: EvoBind2中的AF2 和 AFM 预测中肽链质心(center of mass)之间的偏差,表示两个模型对结构的预测一致性
Cyclic offset 技术
- 支持设计“头尾环化”的环状肽,提高稳定性与细胞膜穿透性
To design cyclic binders, we implement a cyclic offset informing the structure prediction network to connect the peptide amino acids in a continuous cycle. This feature is called the relative positional encoding in the AlphaFold2 and AlphaFold-multimer networks
在 AlphaFold 的结构推理过程中,每个残基对的空间关系都受到“相对位置信息”影响。作者在这个过程中引入了一个“环化偏移量(cyclic offset)”,让网络认为序列的起始和终止残基是邻近的,即构造出“闭环”的结构。
四、实验验证与结果
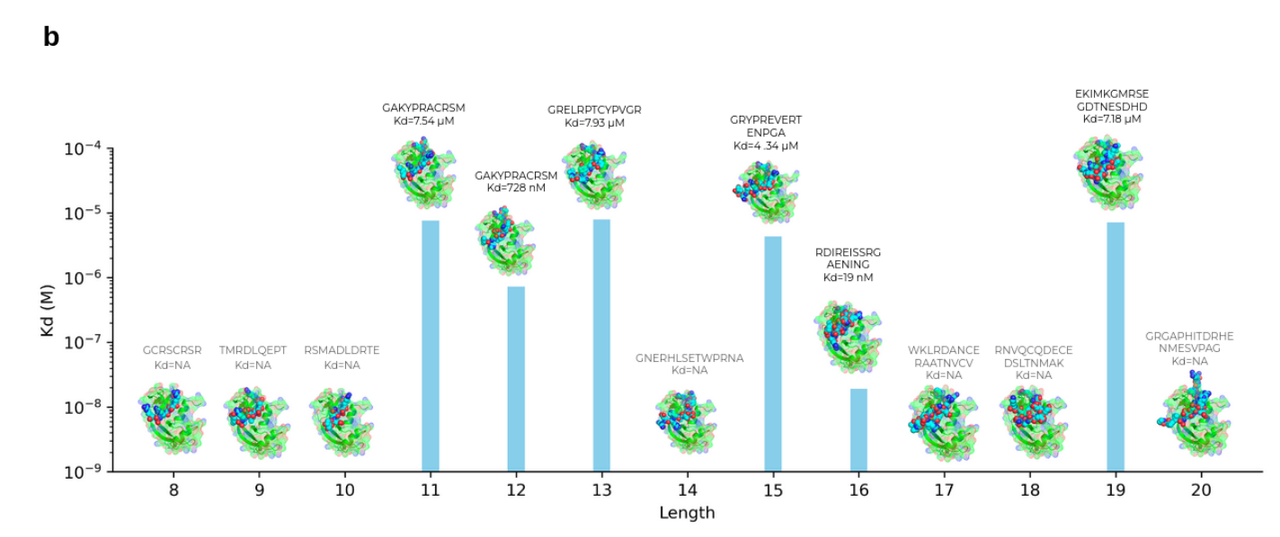
1. 实验设置
靶蛋白:半合成核糖核酸酶(PDB ID: 1SSC),具有已知的肽结合能力
设计并合成13种不同长度(8–20残基)的线性肽,每个长度选取 一个得分最优的序列。
使用SPR(表面等离子共振)测试亲和力,使用 Biacore 8K 设备进行结合动力学测试,测得结合常数 $$K_d$$。
2. 实验结果
| 项目 | 数值/说明 |
|---|---|
| 测试的线性肽数量 | 13条 |
| 显示有效结合的肽 | 6条(约46%成功率) |
| 最强结合肽Kd | 19nM (远低于正对照的35nM) |
| 最弱有效肽Kd | 7.93uM |
| 正对照肽Kd(已知序列) | 35nm |
🔍 附加实验:对抗性验证
比较 EvoBind2 与 AlphaFold-Multimer 预测不一致的序列(疑似“欺骗性设计”):
仅有 2/13 条对抗性序列显示结合;
与此同时,在 EvoBind2 与 AFM 均同意的设计中,成功率是 6/13;
说明引入 AFM 检查可 将成功率提高约三倍。
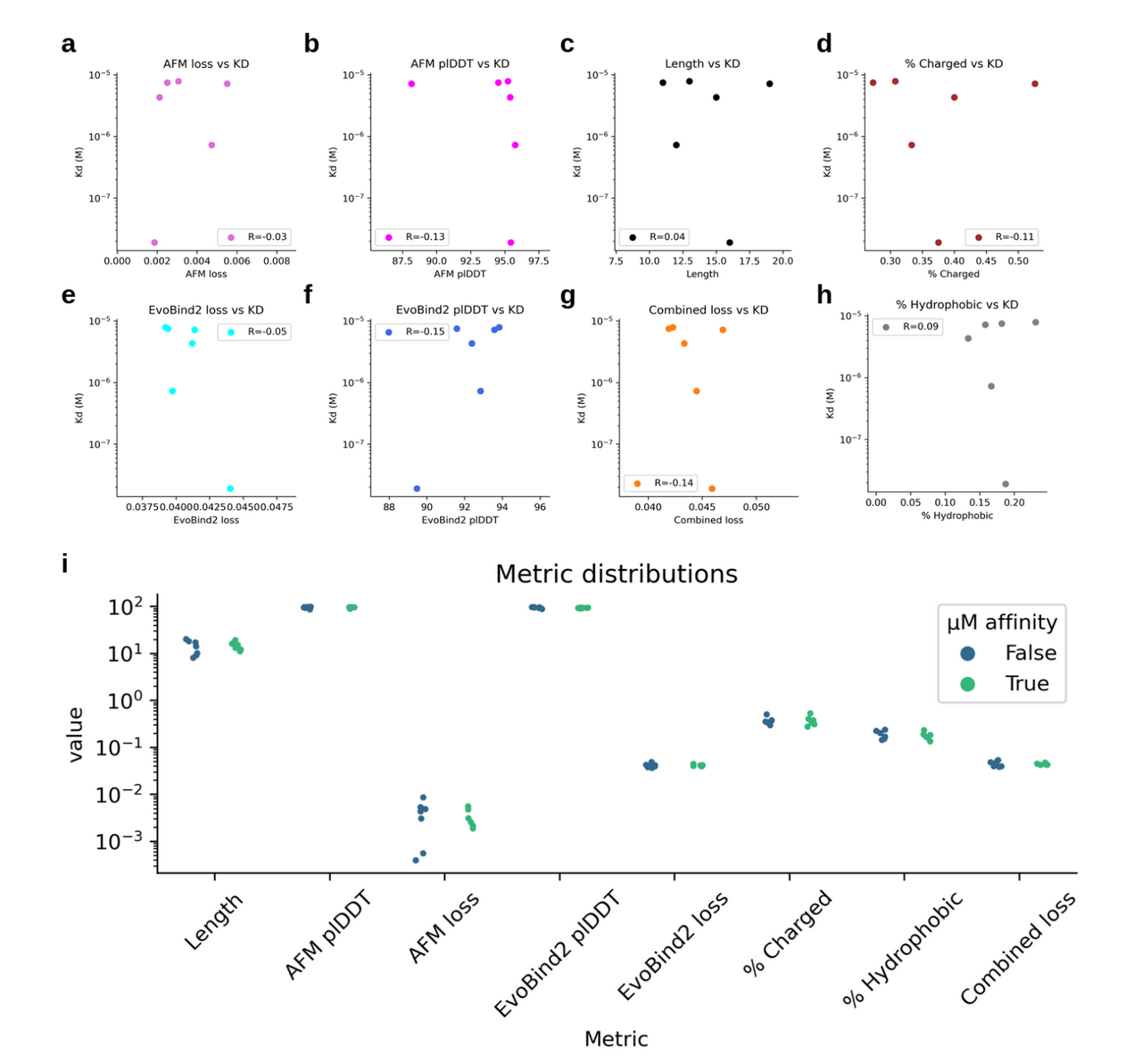
环状肽初步结果
使用相同输入设计环状肽,结构预测成功
尚未进行实验验证,但理论上可行
五、关键结论与意义
EvoBind2能从纯序列信息设计出功能性结合肽,无需结构模板或靶点知识
提供了一个快速、高效、可扩展的蛋白靶点设计方案
对抗性验证机制将设计成功率提升至3倍(从约15%提高到46%)
为新药开发、生物探针设计、蛋白功能研究提供了新途径
六、局限与展望
当前限制:
无法通过预测分数精确区分nM和μM级亲和力
仍需实验验证环状肽功能
未来方向:
融合更多物理-化学特征提升预测准确度
推广至复杂靶标、膜蛋白、多肽复合物等
延伸阅读
本文属于 AI4S文献 栏目。
返回 AI4S文献 → 去公众号阅读完整版 →