BindCraft:一站式设计功能性蛋白 Binder 的自动化平台

今天要介绍的这篇论文《BindCraft: one-shot design of functional protein binders》提出了一个自动化蛋白质结合剂设计平台——BindCraft,它通过反向优化 AlphaFold2 模型,无需高通量筛选即可快速设计出高亲和力的功能性结合蛋白。
在蛋白结合剂开发中,传统方法往往依赖昂贵的实验筛选和结构信息,效率低、门槛高。BindCraft 的出现,为缺乏结构基础和筛选平台的实验室提供了一种“开箱即用”的解决方案,极大降低了蛋白设计的技术门槛,在免疫调节、变应原阻断、CRISPR干预等多个领域展现出广泛应用潜力。
🧠 研究背景与目标
背景:
蛋白-蛋白相互作用(PPIs)在几乎所有生命过程(如免疫、信号转导)中都至关重要。
传统设计蛋白结合剂的方法(如抗体筛选、定向进化等)成本高、效率低。
深度学习模型(尤其是 AlphaFold2)的发展为结构预测与结合设计提供了强大工具。
目标:
开发一种高效、自动化的蛋白质设计平台 BindCraft;
利用 AlphaFold2 模型进行结构预测与反向优化,无需已知结合位点;
实现“设计一次即成功”(one-shot design),降低实验依赖;
推动蛋白质工程向广泛可及、按需定制的发展。
🧪 BindCraft 的核心原理与创新点

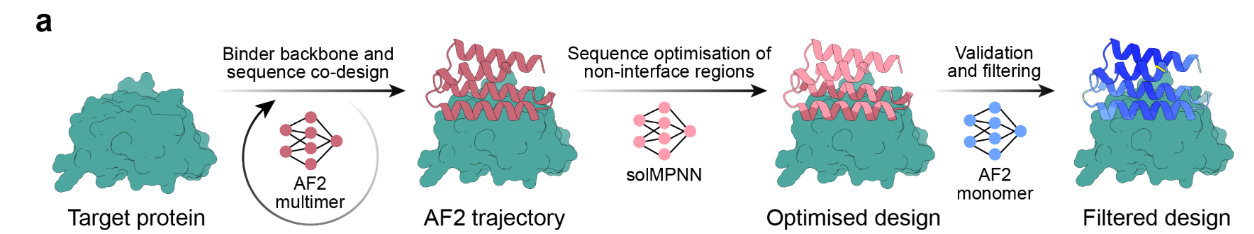
BindCraft 的设计流程如图所示,围绕 AlphaFold2(AF2)展开,形成一个从目标结构输入到功能结合剂输出的全自动闭环系统:
🔁 整体流程概览:
目标蛋白结构输入(Target protein)
用户提供蛋白三维结构(可从AlphaFold、PDB获取);
可选定结合热点(Hotspot)或自动识别结合区域。
结合剂设计初始生成(AF2 multimer trajectory)
采用 AlphaFold2 Multimer 模型进行“幻觉式”设计(hallucination);
结合剂的骨架结构与序列协同优化,同时预测其与目标蛋白的结合界面;
每一步预测都动态调整两者界面结构,提高适配性。
序列微调(solMPNN)
利用 solMPNN 优化非界面区域(即远离结合界面的位置),以提升蛋白的表达性、稳定性和可溶性;
保留关键结合区域不变,从而保持亲和力。
结构验证与筛选(AF2 monomer)
使用 AF2 单体模型重新预测优化后的设计结构;
筛选步骤结合 AlphaFold confidence score 与 Rosetta 打分机制,确保结构稳定且合理;
最终输出具有高亲和力和良好结构质量的结合剂候选。
🌟 关键创新点
反向传播式结构设计:通过 AlphaFold2 模型权重实现反向优化,无需大规模结构采样或数据库模板;
多步神经网络协同优化:将 AF2、MPNN、Rosetta 等模型有机集成,每步优化不同目标(结合位点、稳定性、全局折叠);
非依赖已知位点:无需已知结合区域也能生成高亲和力结合剂,扩大适用范围;
一步到位式(One-shot)策略:不同于传统方法需筛选数千个设计,BindCraft 常仅需十几个设计即有成功结合结果;
普适且自动化:支持多种蛋白类型(如受体、变应原、核酸酶等),流程自动,适合非结构生物学背景的用户。
🔬 实验验证
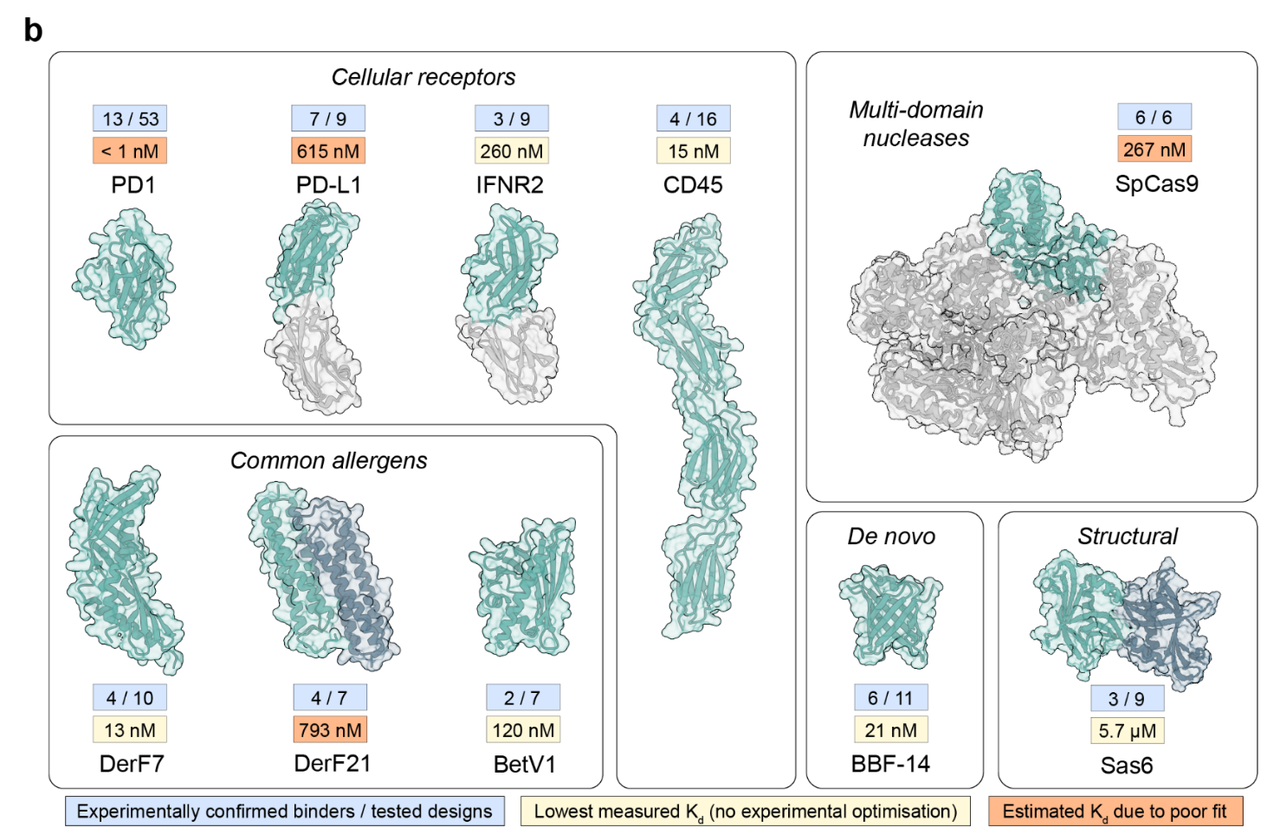
作者通过设计并测试共九个蛋白靶点(共四类蛋白)目标,验证了 BindCraft 在多样场景下的有效性(见上图):
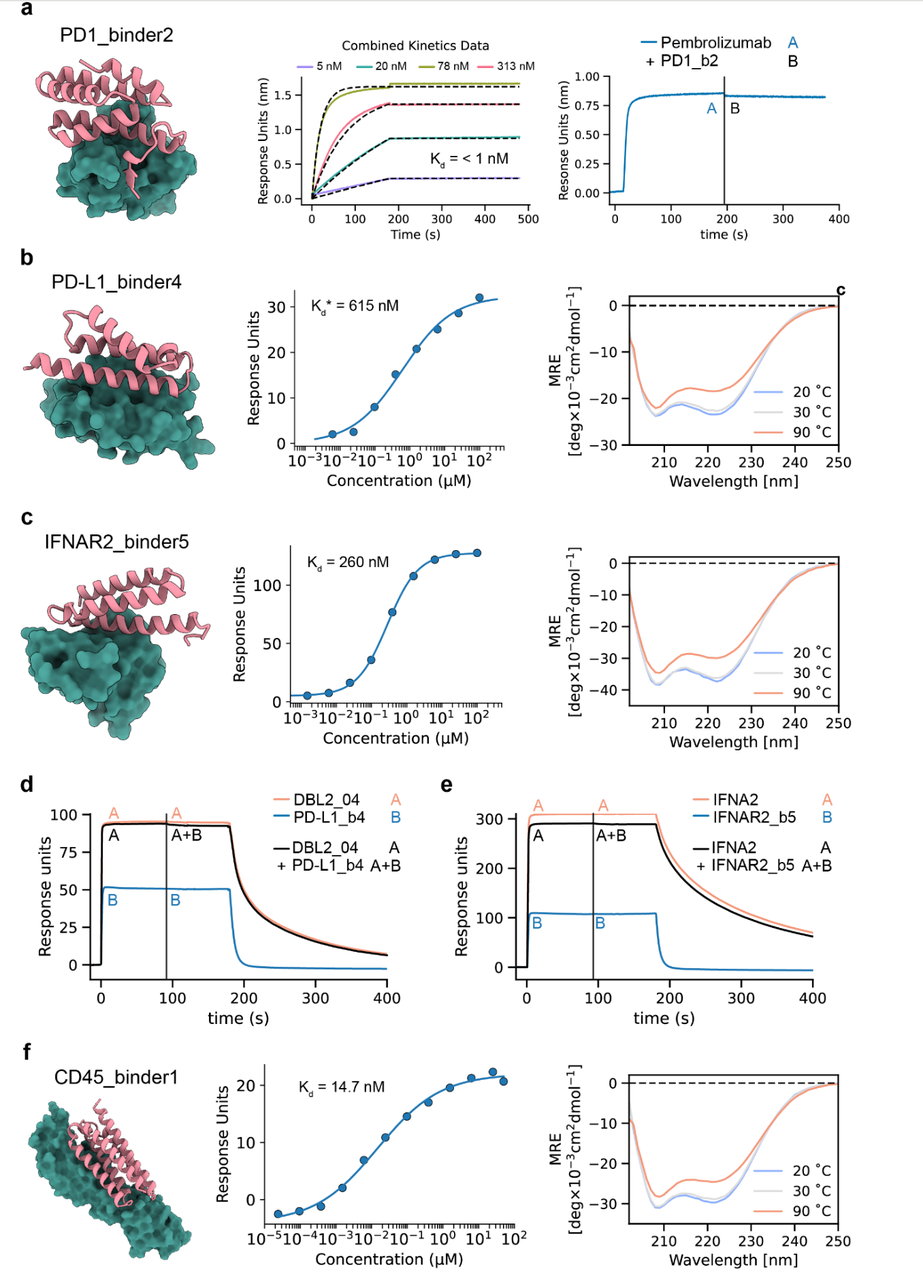
- 细胞受体类(Cellular receptors):如 PD-1、PD-L1、IFNAR2 和 CD45,设计结合剂展示出纳摩尔级亲和力,其中 PD-1 的最佳结合剂亲和力甚至低于 1 nM。
- 常见过敏原(Common allergens):针对 DerF7、DerF21 和 BetV1 的设计表明,BindCraft 可用于阻断 IgE 结合位点,具有潜在治疗价值。

- 多结构域核酸酶(SpCas9):6 个设计中全部验证成功,证明该方法可用于调控基因编辑工具。

- 新设计蛋白与复杂结构蛋白(De novo & Structural):如 BBF-14 和 Sas6,也获得良好结合效果,显示该方法对未知或难靶蛋白也适用。

🧪 BindCraft设计流程简介(Design Protocol)
该流程是一个自动化的设计管线,分为如下关键步骤 :
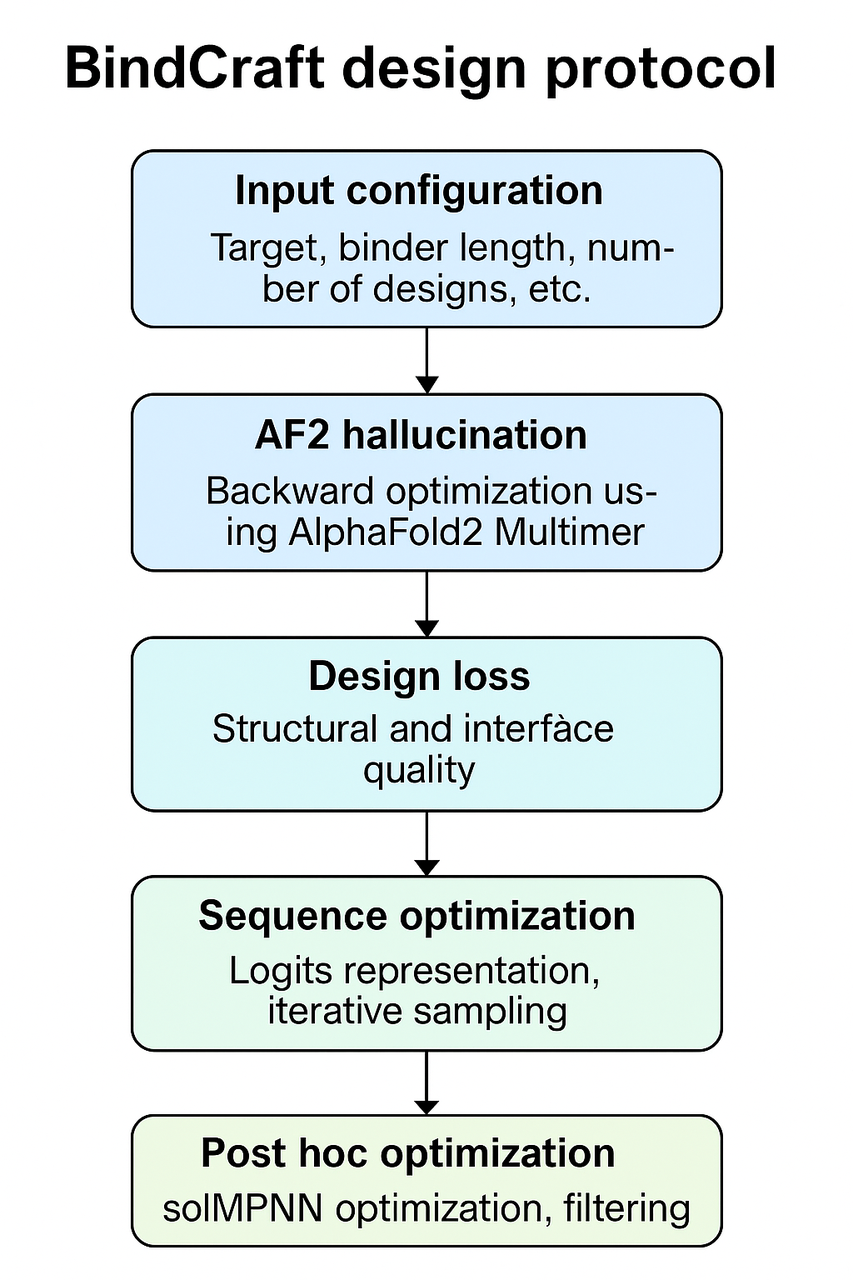
1. 输入配置
用户以 JSON 格式提供目标蛋白(PDB格式)、binder长度范围、目标设计数量等;
可指定结合热点(residue/chains)或不指定,由系统自动选点。

2. AF2幻觉设计(Hallucination)
利用 ColabDesign 实现的 AlphaFold2 Multimer 进行 binder 序列与结构生成;
初始化为随机序列,通过反向传播和误差梯度(L×20)优化每个残基;
使用五种 AF2 模型轮换,提高鲁棒性、避免过拟合。
3. 设计损失函数(Design Loss)
综合多个结构与界面质量指标,常用的权重包括:
结构可信度(Binder confidence pLDDT, weight 0.1)
相互作用界面可信度(interface confidence i_pTM, weight 0.05)
对齐误差 (Normalized prediceted alignment error pAE)等(binder 内部误差: weight 0.4,binder-target间误差, weight 0.1)
接触损失(Reisude contact loss , weight 1.0)
螺旋化倾向(helicity loss, weight -0.3)鼓励非螺旋结构
结合点收缩(终端靠近, weight 0.1)
4. 序列优化阶段(4个阶段)
序列优化阶段是 BindCraft 设计流程的关键环节之一,目标是在已初步拟合目标蛋白的结合剂结构基础上,进一步优化其序列,以提高亲和力、结构稳定性以及生物可表达性。主要通过以下四个阶段来实现:
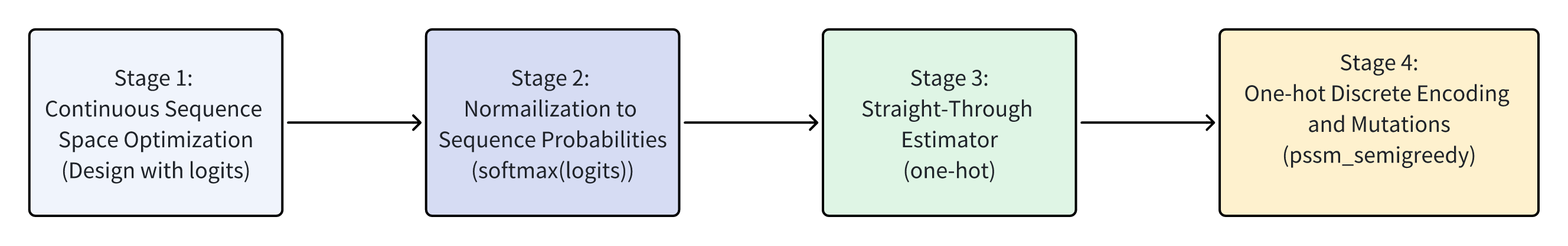
阶段一:连续序列空间优化
目标:探索多样性的序列-结构空间,尤其是在结合界面。
方法:
序列以 logits(连续值)表示,每个位置允许多个氨基酸选择;
每一步的序列计算为:
$$
\text{Sequence} = (1 - \lambda) \cdot \text{logits} + \lambda \cdot \text{softmax}(\text{logits} / T)$$
其中:
$$
\lambda = \frac{\text{step} + 1}{\text{iterations}}$$
$$
T = 1.0$$,为恒定温度
通过迭代,动态地将优化从logits逐步引导至概率分布。
特殊处理:
若 AlphaFold2 confidence 分数(pLDDT)低,提前终止该设计轨迹;
若检测到 β-sheet 构象,增加 AF2循环次数从1到3,提高结构预测精度
迭代轮次:
初始50步用于粗略收敛;
若表现良好,再进行25步延伸优化。
阶段二:softmax 收敛收束
目标:从概率表示过渡到更可信、收敛的氨基酸选择。
方法:
logits → softmax,得到明确的概率分布;
温度 T 随迭代逐步降低,引导从探索向收敛过渡
$$
T = 10^{-2} + (1 - 10^{-2}) \cdot \left(1 - \frac{\text{step} + 1}{\text{iterations}}\right)^2$$
- 学习率也随温度同步衰减,确保收敛稳定。
阶段三:Straight-through Estimator
目标:从概率向离散表示过渡,同时保持可反向传播。
方法:
序列以 one-hot 形式呈现给模型(即每个位点为单个氨基酸);
但反向传播依然通过 softmax 进行(称为“Straight-through”);
实现“看起来是离散的、训练时是连续的”效果。
阶段四:离散微调
目标:最终固定具体氨基酸序列,确保最佳的结构质量和接口表现。
方法:
使用 softmax 概率分布对每个位点进行 随机采样突变;
每次采样$$ X = 0.05 \times \text{binder length} $$个残基,保留最优损失组合;
类似于 局部搜索 + 贪婪优化。
✅ 最终筛选标准
pLDDT < 0.7 → 排除;
界面接触点 < 7 → 排除;
骨架严重冲突或异常扭曲结构 → 排除。
5. 后处理优化 (solMPNN + AF2 + Rosetta)
这一阶段的目的是对初步设计的结合剂进行结构精炼和功能验证,最终确保它们不仅“结合得上”,还具备良好的稳定性、表达性和能量学合理性。这是 BindCraft 区别于许多简单AI生成方法的重要一环。
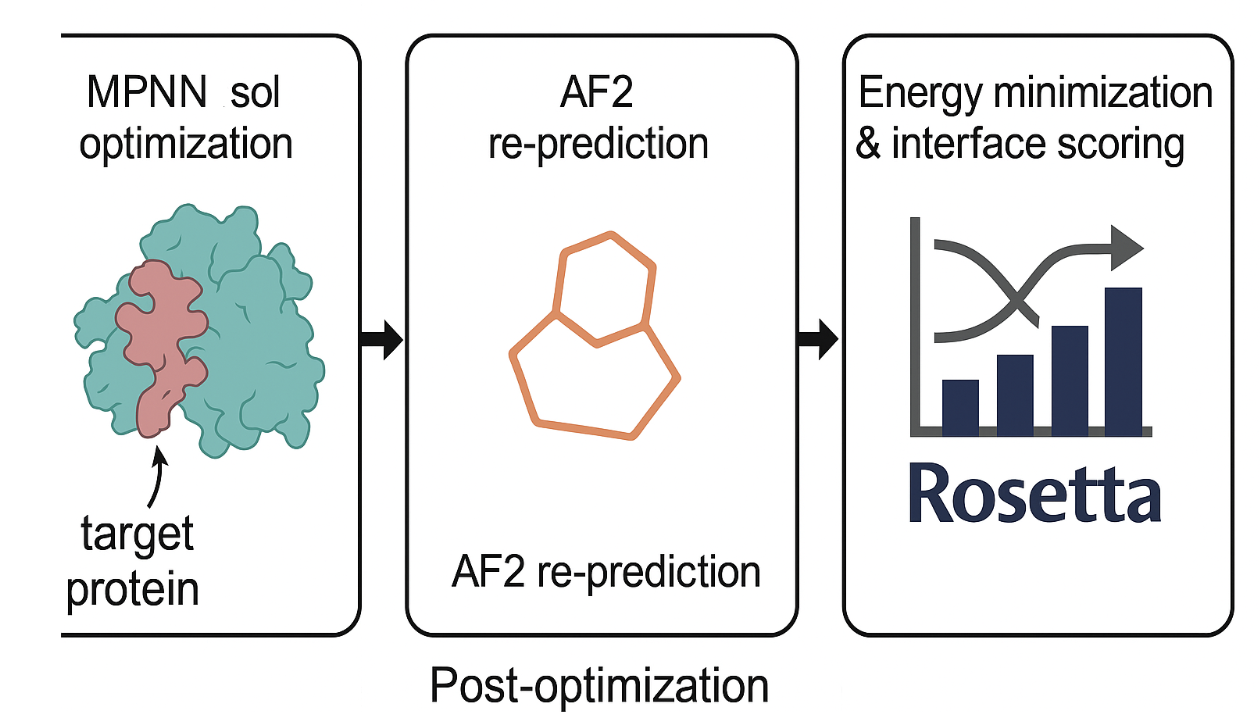
🌟 1. MPNNsol 优化:提高结合剂稳定性与溶解性
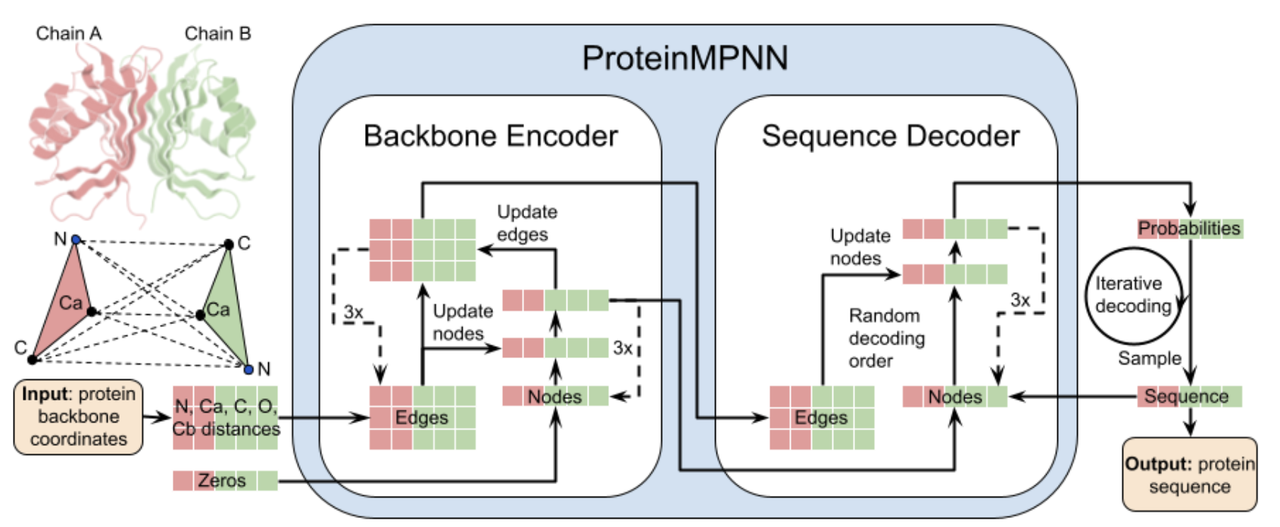
适用于:已通过结构筛选的“成功设计轨迹”;
工具:使用 ProteinMPNN 的溶剂权重版本(MPNNsol);
优化策略:
保留结合界面附近4 Å以内的残基,避免破坏亲和力;
对其余 核心和表面残基重新设计,每个轨迹生成 20 个新序列;
温度设为 0.1(控制采样多样性),不引入骨架噪声(backbone noise = 0.0)
🔁 2. 结构验证:AF2单体模式再预测
使用 AlphaFold2 monomer 模型对新序列进行重构预测;
参数设置:
循环(recycle)次数为3次,提高预测精度;
使用 2个基于模板的模型,但以“单序列模式”运行,防止多序列比对偏差;
目的:验证MPNNsol优化后的序列是否保持结构可靠性。
⚙️ 3. 能量最小化与界面评估:Rosetta分析
使用 Rosetta 的 FastRelax 模块对 AF2 预测结构进行全原子能量最小化(200次迭代);
再通过 InterfaceAnalyzer 模块分析蛋白-蛋白界面质量,计算包括:
界面能量(binding energy);
接触面积;
副链/主链结构调整对结合效果的贡献;
6. 筛选标准总结:确保设计质量的过滤条件
在 BindCraft 流程的最后阶段,所有设计将通过一系列预设过滤条件进行筛选,以确保仅保留结构可信、功能合理、具实验潜力的高质量binder。
这些标准基于既往文献经验并在本研究中进一步优化,具体包括:
AF2整体结构可信度(pLDDT):> 0.8
AF2结合界面可信度(i_pTM):> 0.5
AF2界面对齐误差(i_pAE):> 0.35(值越高表示对齐误差越小)
Rosetta界面形状互补性(Shape Complementarity):> 0.55
未饱和氢键数(unsatisfied H-bonds):< 3
结合剂表面疏水性:< 35%
结合态与游离态的RMSD差异:< 3.5 Å
这些标准共同确保输出binder结构稳定、界面紧密、可溶性良好、预测结果可信,适合进入实验验证阶段。
🧾 总结与讨论
🔬 1. 背景与意义
蛋白-蛋白相互作用(PPIs)的计算设计长期是蛋白质工程的核心挑战;
难点在于:我们对分子识别机制(特别是结合位点的物理化学特征)仍了解有限;
借助 AlphaFold2 等深度学习结构预测工具,现已能对新设计的结合剂进行更准确评估。
⚙️ 2. BindCraft 的独特优势
基于 AF2 网络的反向传播优化,首次实现结合剂“幻觉式”设计;
相比于多数固定靶点的方法,BindCraft 允许目标蛋白本身存在柔性,有助于捕捉结合诱导的构象变化;
系统可自动识别高结合倾向位点,无需事先定义目标区域。
📈 3. 实验表现与成功率
在12种复杂目标蛋白上设计结合剂,亲和力大多达到纳摩尔水平,极少数为微摩尔;
平均成功率为 46.3%,显著高于传统方法(如 RFdiffusion、AlphaProteo);
在社区挑战中设计的结合剂还获得第一名,成功靶向 EGFR 高难度靶点。
🔍 4. 面对的挑战与扩展前景
PPI设计的主要挑战在于选择合适的结合表位,尤其是疏水且结构平坦的区域;
BindCraft 能处理既有已知结合位点的蛋白,也能在 de novo、变应原或核酸酶等复杂系统中自动定位结合区域;
在设计结合 蛋白-核酸界面结合剂方面也展现出潜力,为未来开发转录因子抑制剂等功能工具提供新方向。
🧪 5. 局限性与潜在问题
参数需个性化调整
默认设置(迭代次数、损失权重、筛选阈值)可能不适用于所有目标蛋白;
对于不同靶标,可能需要根据经验调整这些参数以获得理想结果。
结合位点选择仍关键
尽管 AlphaFold2 在自动识别结合位点方面表现优秀,指定热点位点仍能提升设计成功率;
自动识别虽然有效,但在特定生物功能需求下,人工选择更具可控性。
亲水界面预测较弱
- 相比疏水界面,AF2 在亲水性结合界面的预测与设计效果更差,需谨慎解读结果。
结构变形常见但可控
在设计过程中,某些结合剂轨迹可能出现结构变形或“被压扁”(squashed);
这是 AF2 Multimer 对输入序列高度敏感所致,属正常现象;
此类无效轨迹会被系统自动识别并快速剔除,不影响整体流程稳定性。
因此,使用 BindCraft 时要注意:虽然系统自动化程度高,但仍需人为判断与适配,特别是在新靶点上使用时。
📚 原文与资源信息
论文标题:BindCraft: one-shot design of functional protein binders
发表平台:bioRxiv(预印本)
原文链接:https://www.biorxiv.org/content/10.1101/2024.09.30.615802v3
代码仓库(GitHub):
BindCraft 官方开源代码与使用说明已公开,支持用户在本地运行完整设计流程:
延伸阅读
本文属于 AI4S文献 栏目。
返回 AI4S文献 → 去公众号阅读完整版 →