3,766 个实验数据揭示 Binder 成功率的关键指标
今天要和大家分享一篇刚刚上线的 bioRxiv 预印本 ——
《Predicting Experimental Success in De Novo Binder Design: A Meta-Analysis of 3,766 Experimentally Characterised Binders》(2025.08.14 发布)。
为什么值得聊这篇文章?有三个原因:
规模前所未有:它汇总了 3,766 个经过实验验证的 de novo binder 设计,涵盖 15 个多样化的靶点,是目前最大规模的数据集。
方法学突破:作者不仅比较了 AF2、AF3、Boltz-1 等预测工具,还提出了一个新的关键指标 —— AF3 ipSAE_min,在 binder 筛选上远胜现有方法。
实用价值高:文章最后给出了可直接落地的筛选规则,对所有做 binder 设计的人都有立刻能用的指导意义。

一、开篇引入
在过去几年里,de novo 蛋白质 binder 设计(从零开始设计与目标蛋白结合的小分子蛋白)逐渐成为结构生物学和合成生物学的热门方向。随着 RFdiffusion、BindCraft、AlphaProteo 等方法的出现,我们已经能在计算机上快速生成成千上万的候选设计。
然而,真正能在实验中“成功结合”的比例却依旧很低。原因很简单:实验验证昂贵、耗时,而 in silico 过滤手段还不够精准。大家常用的 AlphaFold2 (AF2) 预测信心指标(比如 ipAE、ipTM)确实能在一定程度上提升命中率,但远远谈不上“稳定可靠”。
于是,一个关键问题摆在研究者面前:
👉 我们能否找到更稳健、跨靶点通用的预测指标,用来提前筛掉失败设计?
这正是本文《Predicting Experimental Success in De Novo Binder Design》试图回答的问题。作者们不仅汇总了一个 3,766 个设计、15 个靶点的前所未有的大数据集,还系统性比较了 AF2、AF3、Boltz-1 等结构预测工具输出的各种特征。最后,他们提出了一套简单、可解释、但十分有效的 binder 过滤策略。
这篇文章的最大价值在于:它让 binder 设计的“成功率预测”第一次有了大规模的系统 benchmark,并且给出了能直接应用在实际工作中的筛选规则。
二、数据与研究设计
要想真正回答“哪些 binder 会在实验里成功”这个问题,必须有足够大、足够多样化的数据。
作者们这次汇总了一个前所未有的 dataset:3,766 个经过实验验证的 binder 设计,涵盖了 15 个结构和功能各异的靶点。其中不仅有经典的受体酪氨酸激酶(EGFR、FGFR2、TrkA 等),也包括免疫相关分子(IL2Ra、PDL1)、病毒抗原(SARS-CoV-2 RBD),甚至还有蛇毒神经毒素和 pMHC 复合物。
这一大杂烩式的 dataset,有几个值得注意的特点:
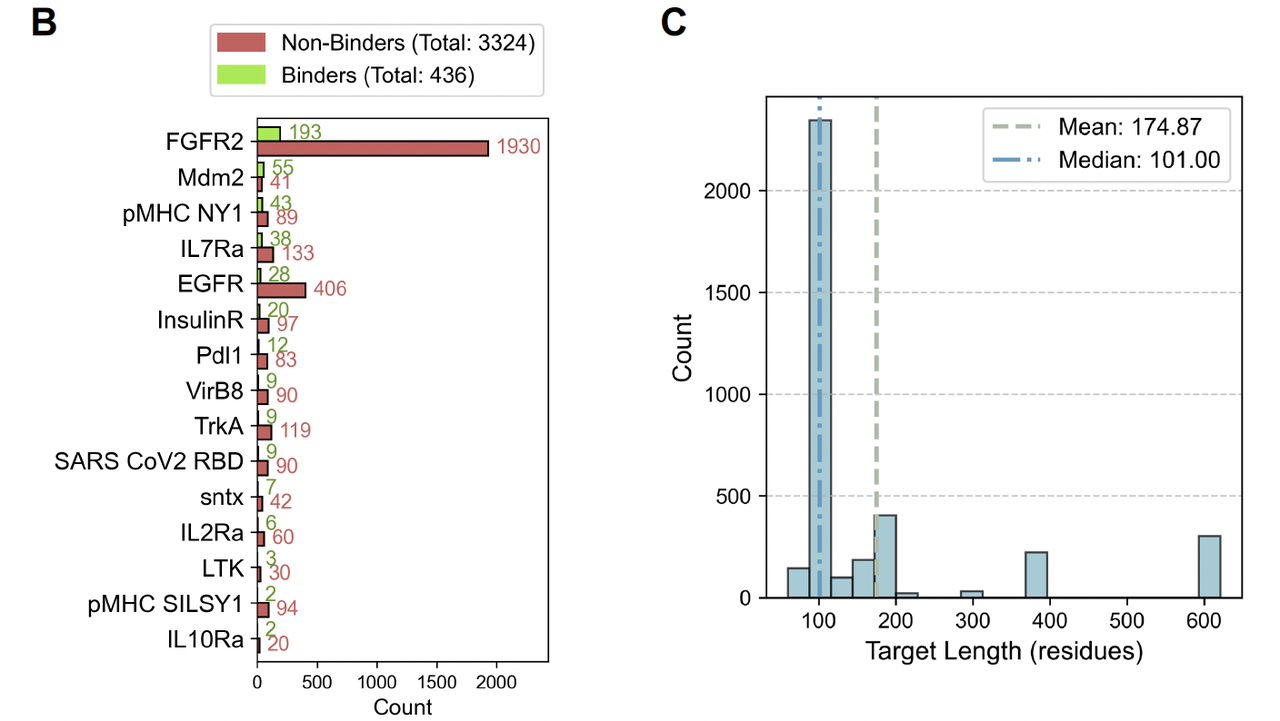
Binder 成功率很低:3766 个设计里,只有 436 个(11.6%)在实验中被验证为有效。
数据严重不平衡:有的靶点几乎全是失败设计,有的则 binder 相对较多。
靶点差异极大:从 60 个氨基酸的小毒素,到 600 多个氨基酸的受体,跨度很大。
这些特征在 Figure 1 里被清晰地可视化:
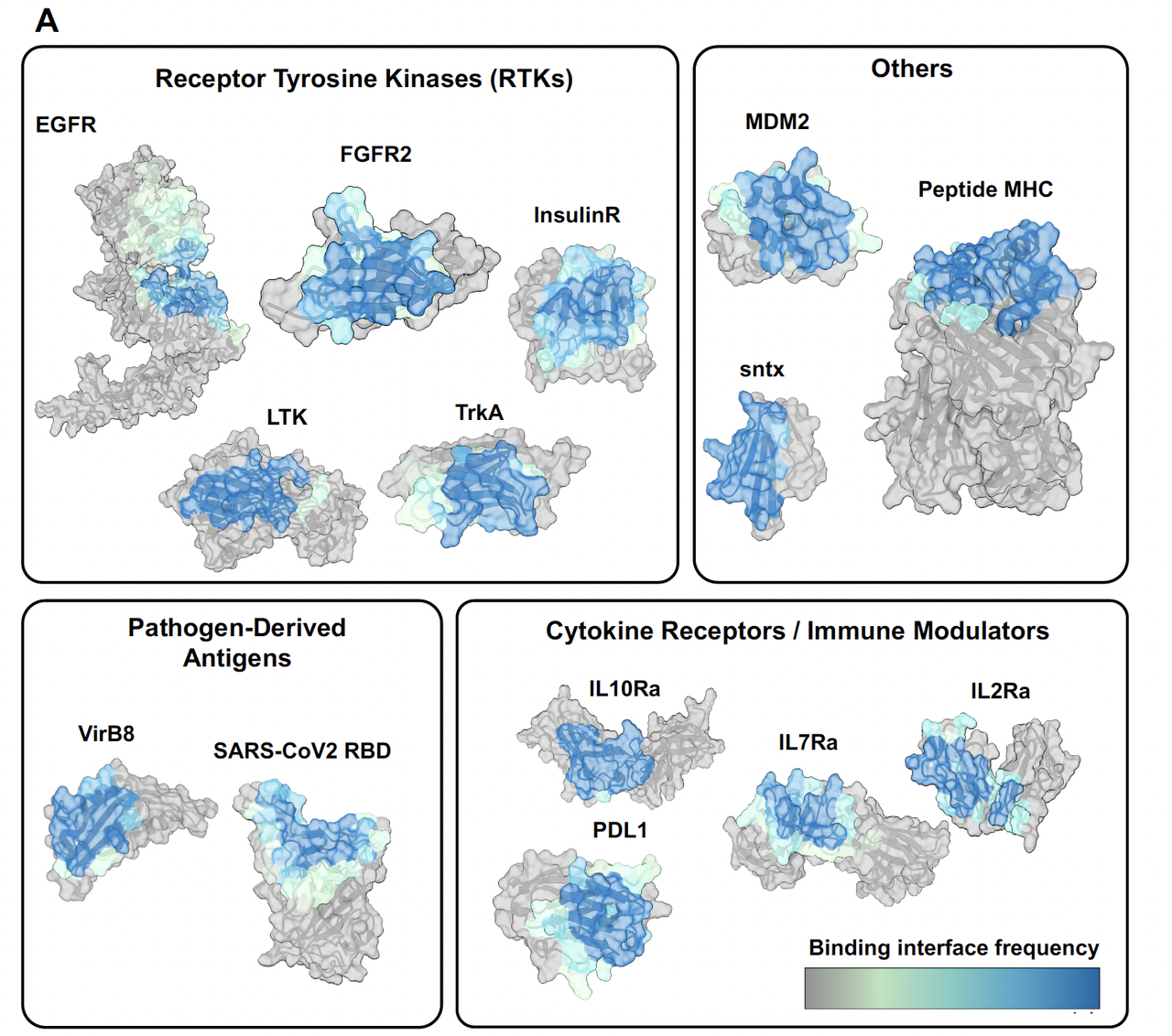
Figure 1A 展示了所有 binder 的结合位点分布,可以看出不同靶点上的真实 binder 都倾向于结合在一些高度保守的界面区域。
Figure 1B 是一个直观的统计:不同靶点的正负样本差异悬殊,比如 FGFR2 有大量设计和验证,而像 LTK 这样的靶点几乎没几个成功案例。
Figure 1C 则画出了靶点长度的分布,说明数据集覆盖了广泛的结构空间。
📌 这一部分的意义在于:
数据规模足够大,保证了结果的统计学可靠性;
数据的多样性和不平衡性,也让预测问题更具挑战性,更接近真实科研环境。
换句话说,这个 dataset 本身就已经是一个领域级的资源,不仅能用来做本文的分析,还可以成为未来 benchmark 的基础。
三、计算 Pipeline 的优化
在拥有了一个覆盖 3,700 多个 binder 的大数据集后,下一个问题是:如何高效地对这些设计进行结构预测和特征提取?
要知道,常规的 AlphaFold 或 Boltz-1 预测,一个复合物可能要跑几十分钟甚至更久。对于几千个设计,这几乎是不可完成的任务。
作者的解决方案是——开发一个统一的自动化 pipeline,能够把所有候选 binder–target 复合物,依次送入不同的预测工具(AF2 初始 guess、ColabFold、Boltz-1、AF3),并在输出结构的基础上提取超过 200 种特征(包括结构、能量、置信度和序列信息)。
关键优化点:
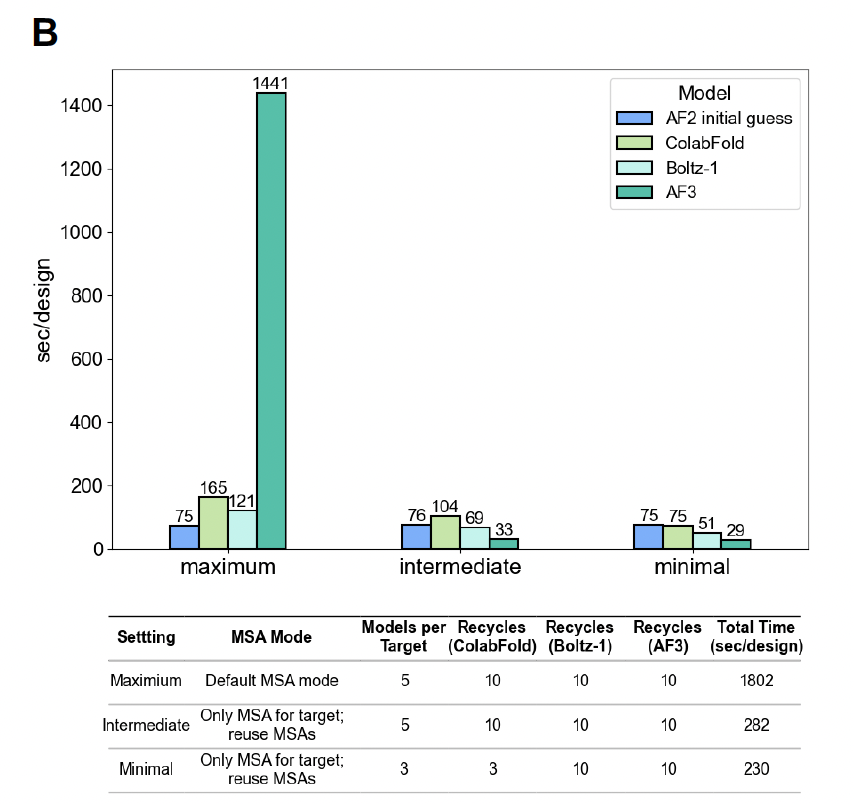
他们特别测试了不同运行配置(最大化配置 / 中间配置 / 极简配置)的速度和准确性差别。
速度提升:
在“极简配置”下,平均预测时间从 1800 秒降到 230 秒,提升了 87% 的效率。
AF3 的加速尤为明显,从 1441 秒缩短到 29 秒!
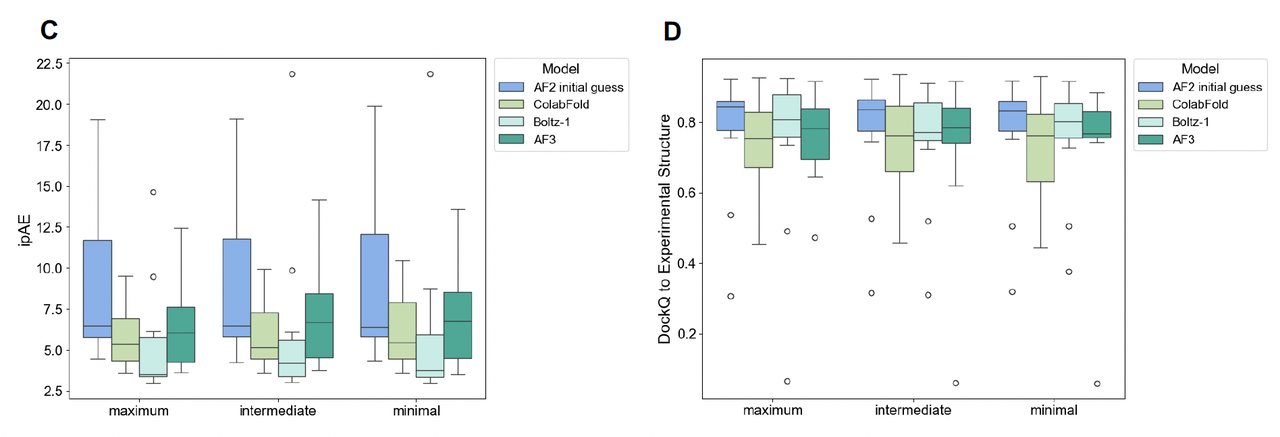
准确性保持:
Figure 2C/D 展示了不同配置下的模型信心指标(ipAE)和与真实结构的 DockQ 对比。
结果表明,即便是最轻量级的配置,预测质量几乎没有下降。
📌 Figure 2 的逻辑:
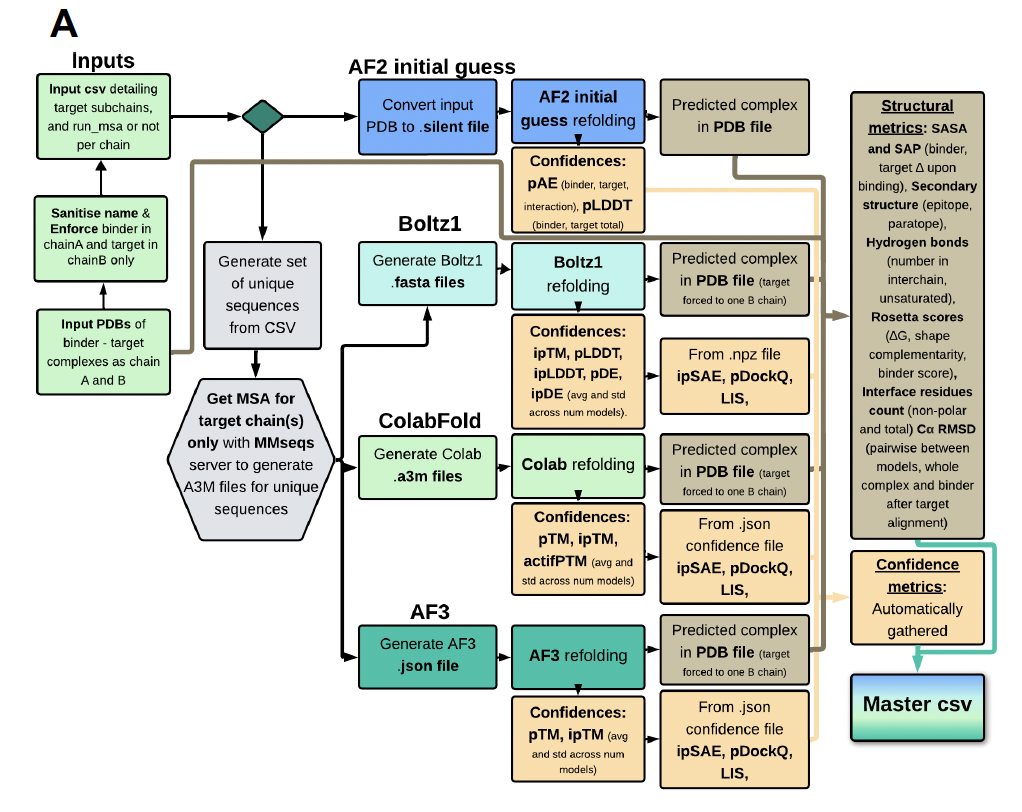
Figure 2A:pipeline 的整体架构 → 统一输入、统一输出、全自动化。
Figure 2B:不同配置下的耗时比较 → 极简配置最快。
Figure 2C/D:质量评估 → 即便是极简配置,预测结果依旧可靠。
意义
这一部分告诉我们:
作者不仅仅是做了个 meta-analysis,而是真正解决了规模化预测的计算难题;
得益于 pipeline 的优化,他们才能在后续系统性比较不同指标的预测能力;
对社区而言,这个 pipeline 也将成为一个可复用的 标准化工具。
四、哪些特征真正预测结合?
在建立了统一 pipeline 后,作者开始回答最关键的问题:哪些指标能区分成功的 binder 和失败的 binder?
- 单一特征的比较
结果非常明确:
AF3 的 ipSAE_min 是最强的单一指标。
它在平均精度(AP)上,比传统常用的 AF2 ipAE 提升了 1.4 倍。
为什么 ipSAE_min 更好?
它只关注 高置信度的结合界面残基对,并取两条链预测误差中的“最小值”,更严格地捕捉了“最薄弱的一环”。
这种界面导向的评分方法,比全局性指标(如 ipTM 或 ipAE)更能反映真实的结合。
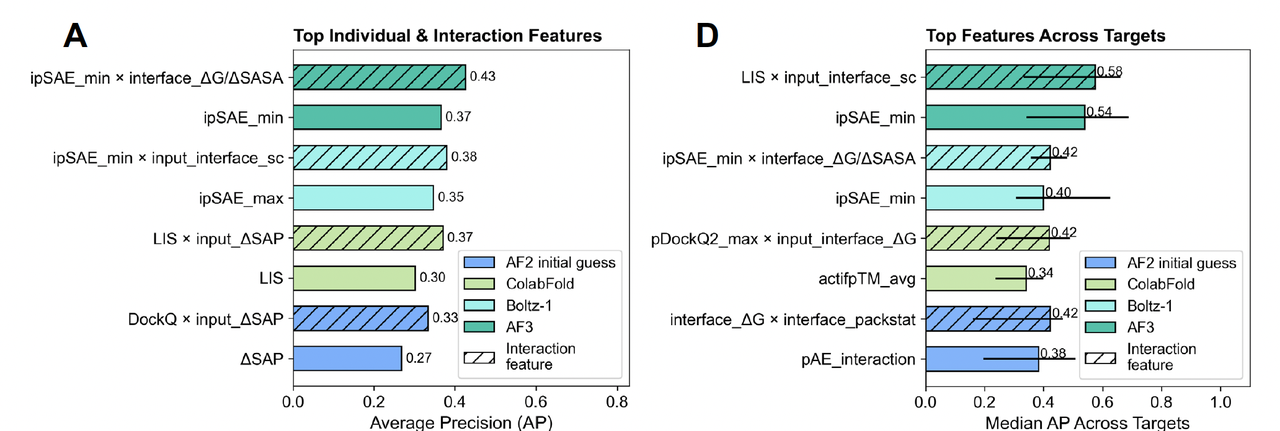
- 特征组合的优势
作者进一步测试了交互特征(把两个指标相乘)。
结果发现,最优组合往往是:
ipSAE_min × 界面 ΔG/ΔSASA(界面能量/界面面积比)
ipSAE_min × shape complementarity(界面互补性)
这些组合反映了一个直观逻辑:结构预测的置信度 + 物理化学界面特征 → 更接近实验真相。
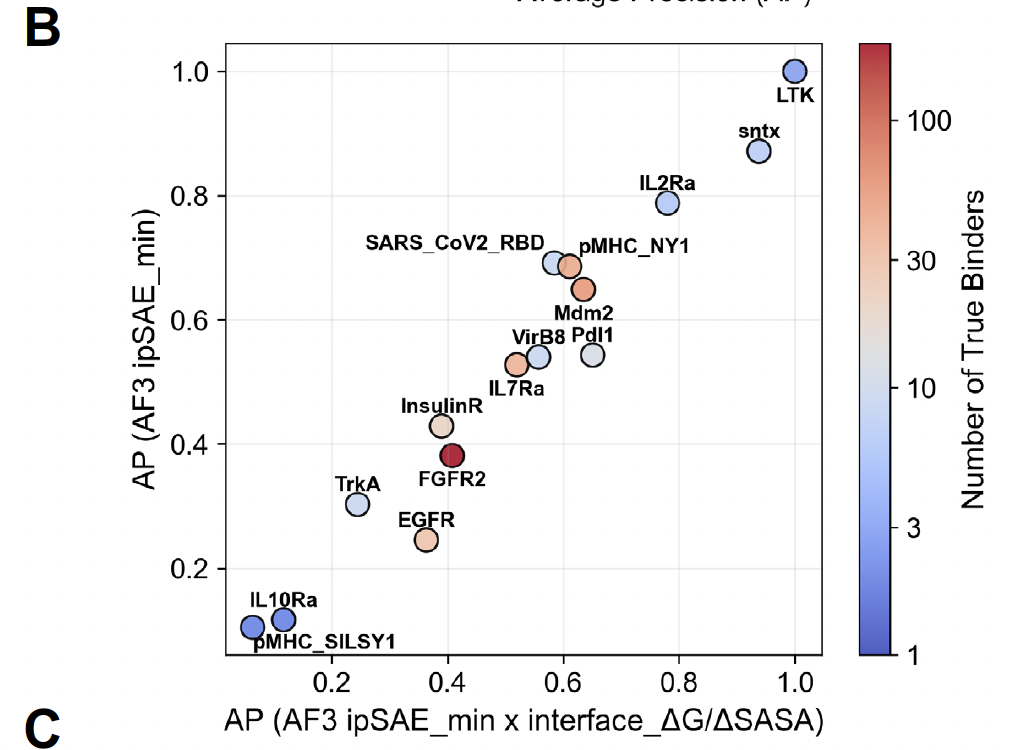
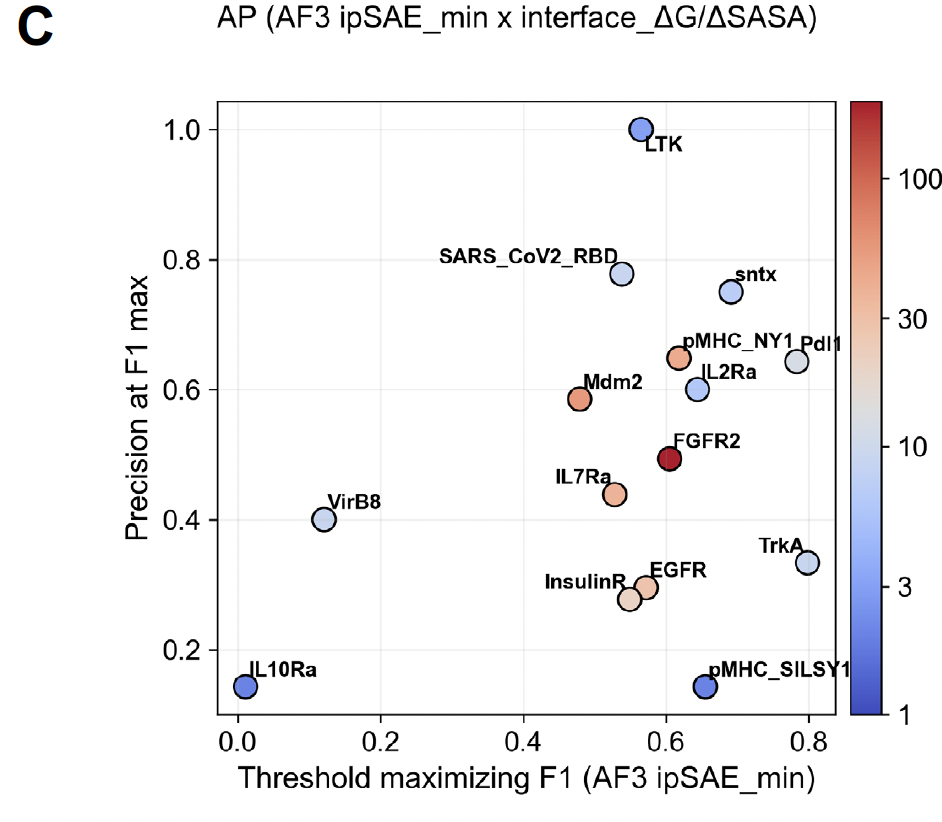
靶点依赖性
不同靶点上的表现差异很大。
比如有的靶点上,ipSAE_min 的 precision 接近 1;但在另一些靶点上则只有 0.1。
这说明:binder 的可预测性是靶点相关的,有些靶点天生更“难搞”。
- 阈值与筛选策略
作者测试了几种实际可用的筛选阈值:
固定 recall (0.2 / 0.4)
F1 最大化的阈值
在几乎所有情况下,AF3 ipSAE_min 及其组合指标都优于 AF2 ipAE 和 AF3 ipTM。
Precision-recall 曲线(Figure 3F)清楚展示了这种优势。
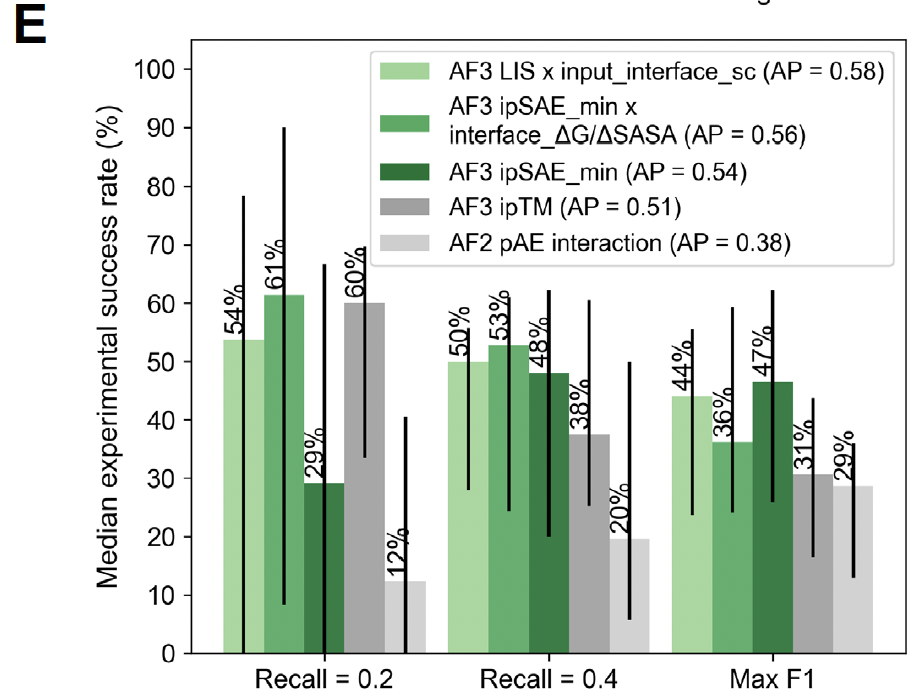
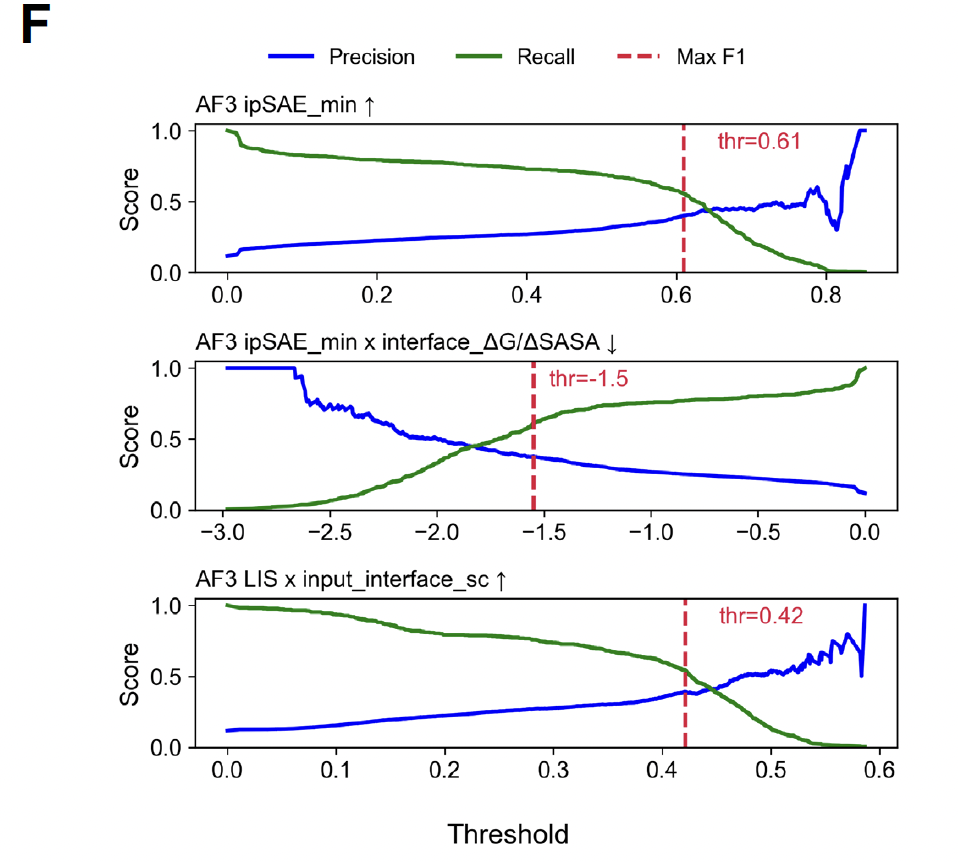
📌 Figure 3 的逻辑线:
Figure 3A:不同单一指标 & 组合指标的 AP 比较,突出 ipSAE_min 最强。
Figure 3B–D:不同靶点上的表现差异,强调预测难度不均。
Figure 3E/F:实际筛选时不同指标的 precision–recall 表现。
总结这一部分
作者得出了一个非常实用的结论:
如果你只能用一个指标,那就选 AF3 的 ipSAE_min。
如果你可以用两个指标,那就加上界面 ΔG/ΔSASA 或 shape complementarity。
换句话说,他们用数据证明了:界面导向 + 物理化学特征的组合,是最稳健的预测方案。
五、特征组合与模型选择
前一部分的分析告诉我们,单一指标(尤其是 AF3 ipSAE_min)已经很强,而与物理化学特征结合后还能更进一步。但在实际应用中,很多人会想:是不是应该用更多特征,甚至上复杂模型(比如深度学习、XGBoost),才能更好?
作者专门做了一个系统比较,答案令人意外却很有价值:
👉 少量特征 + 简单线性模型,其实就够了。
- Greedy 特征选择
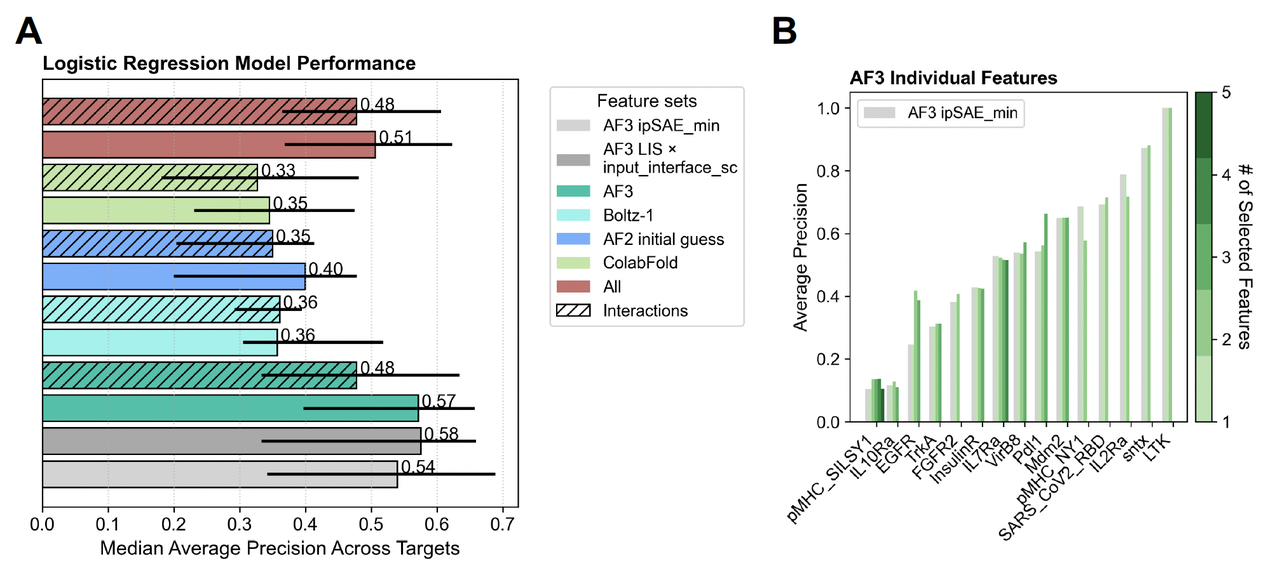
作者采用了 greedy feature selection + logistic regression 的方法:
在 15 个靶点中,逐个留一(Leave-One-Group-Out),保证评估的普适性。
在每一轮里,只保留真正能提升平均精度(AP)的特征。
结果:
平均下来,每个模型只会增加 2–5 个特征,就达到性能上限。
当特征加得更多时,反而因为过拟合而失效。
- 哪些特征最常被选中?
在 AF3 派生的特征中,有几个指标反复被选中:
AF3 ipSAE_min(几乎每次都被选中 → “核心指标”)
RMSD_binder(binder 输入结构与 AF3 预测结构的差异)
input_interface_shape_complementarity(界面互补性)
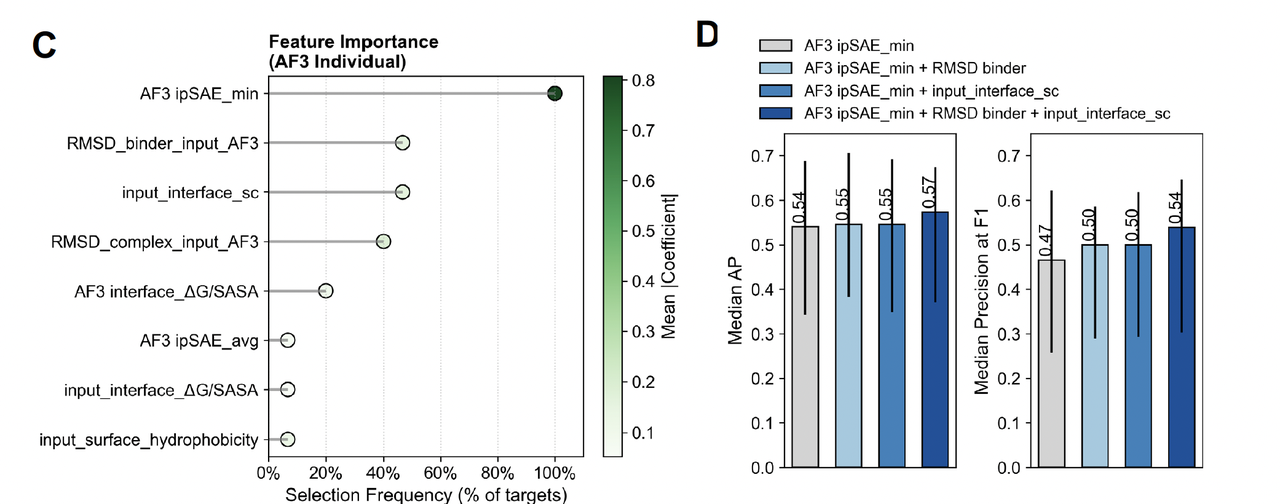
这三者的组合,几乎已经能捕捉到预测的全部信息。
- 实际应用:Top-K 筛选
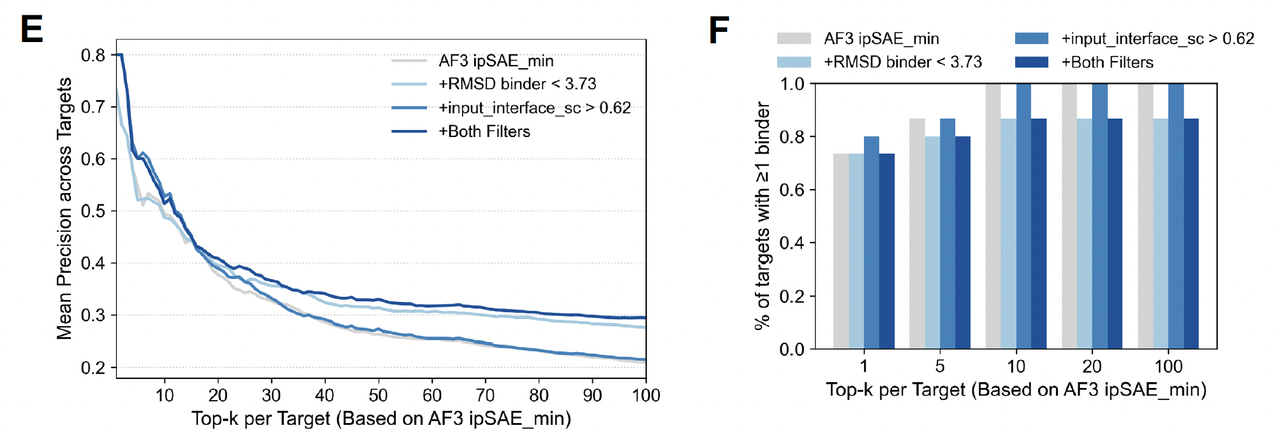
作者还测试了一个贴近现实的场景:
- 假设你每个靶点只能测试 10–20 个候选 binder,能不能用这套指标挑中至少一个真正的 binder?
结果(Figure 4E/F):
只用 AF3 ipSAE_min 排序,就能在前 10 个候选里,几乎保证每个靶点至少有一个真实 binder。
加上界面互补性筛选,precision 在小样本 (K=1–20) 的情况下还能进一步提升。
📌 Figure 4 的逻辑线:
Figure 4A:不同模型的总体表现 → 简单 logistic regression 已经很好。
Figure 4C:哪些特征最常被选中 → ipSAE_min、RMSD_binder、shape complementarity。
Figure 4D:三特征组合的效果最佳。
Figure 4E/F:Top-K 筛选的实际 precision 提升。
总结这一部分
作者给出了一个非常接地气的结论:
binder 成败预测,不需要几十个复杂特征,更不需要黑箱模型。
只要结合 2–3 个关键指标(ipSAE_min + RMSD_binder + shape complementarity),用最简单的线性模型,就能稳定筛出成功率最高的候选。
这意味着,今后的 binder 设计者可以直接把这套“组合规则”嵌入自己的筛选 pipeline,而不用依赖黑箱式的 AI 模型。
六、讨论与应用价值
通过前面的大规模比较和系统分析,作者提出了几条非常有实践意义的结论。
- AF3 指标的优势
AF3 的 ipSAE_min 是目前最稳健、跨靶点最通用的预测指标。
它比 AF2 的 ipAE 提升了 1.4 倍的平均精度,在 binder 设计筛选里完全可以作为新的一代标准指标。
简单比复杂更可靠
复杂模型(比如 XGBoost、多特征堆叠)并没有带来更好的效果,反而容易过拟合。
只需 2–3 个关键特征(ipSAE_min + RMSD_binder + shape complementarity),用线性模型就能实现稳定预测。
这意味着未来的筛选 pipeline 可以 更轻量、更可解释。
应用场景
在实际设计中,研究者往往只能测试少量候选(10–20 个)。
本文证明:只要用 ipSAE_min 排序,就几乎能保证命中一个真实 binder;再加上界面互补性筛选,precision 会进一步提升。
这为实验设计者提供了一个“高性价比”的策略:在有限的实验资源下,提高成功率。
对领域的意义
这不仅仅是一篇算法论文,而是一次方法学标准化的推动。
作者开放了完整 dataset 和 pipeline,未来研究者可以在同一个 benchmark 上比较不同方法,减少“各玩各的”的割裂。
从长远看,这种标准化会显著加速 de novo binder 设计走向成熟应用(如药物研发、诊断工具开发)。
📌 一句话总结本文价值:
这篇工作告诉我们,预测 binder 成败的关键并不在于复杂模型,而在于找到少量真正稳健的指标 —— 其中 AF3 ipSAE_min 是核心,结合界面结构特征即可显著提升实验命中率。
七、结尾:给蛋白质设计者的启发
这篇文章的意义不仅在于提出了新的预测指标,更在于它传递出一个重要信号:
在 de novo binder 设计的赛道上,数据规模化与方法学标准化,正在逐渐取代经验主义。
未来,binder 设计可能会出现几个趋势:
“小而精”的筛选策略 —— 不再依赖海量实验,而是通过 2–3 个关键指标精准命中。
开放数据与社区共享 —— 本文的数据集和 pipeline 将成为公共基准,推动不同方法的公平比较。
跨学科融合 —— 结构预测(AI)、物理化学特征(计算化学)、实验验证(生物学)将进一步整合,形成闭环。
对于正在做 binder 设计的研究者来说,最直接的 takeaway 是:
👉 如果你明天就要筛选新一批 binder,请优先考虑 AF3 ipSAE_min,并结合界面互补性或 RMSD 来过滤。
这套规则简单、可解释,而且已经在 3,766 个设计的实验数据上得到了验证。
或许在不久的将来,我们会看到 binder 设计从“碰运气”走向“可预期”,而这篇文章正是一个里程碑。
延伸阅读
本文属于 AI4S文献 栏目。
返回 AI4S文献 → 去公众号阅读完整版 →