蛋白质 Binder 设计的三代演进:从 Rosetta 到 RFdiffusion
在生物学的世界里,蛋白质是最重要的“分子机器”。它们像乐高积木一样由氨基酸拼装而成,却能完成几乎所有生命活动:从催化反应,到信号传导,再到结构支撑。没有蛋白质,就没有生命。
长期以来,人类在“药物开发”上主要做的事情是 寻找 蛋白质,而不是 创造 蛋白质。我们要么依靠大自然里已经存在的分子(比如抗体、激素、毒素),要么通过大规模筛选找到有用的片段。但这种方式有两个根本性问题:
效率低:像大海捞针一样,需要成千上万次实验,耗时耗力。
局限大:我们往往只能找到“差不多能用”的分子,而不是“完全为我们量身定制”的分子。
想象一下,如果你需要一把钥匙来打开某个复杂的锁(比如癌细胞上的受体蛋白),传统方式就是到处翻找已有的钥匙,试一把算一把。但如果可以 直接设计一把全新的钥匙,精确匹配这把锁呢?这就是 蛋白质设计(protein design) 的愿景。
几十年来,这个愿景听上去更像科幻小说,因为蛋白质折叠与结合的规律极其复杂,远超人类直觉。直到最近几年,随着 计算方法与人工智能的飞跃,我们才真正走到可以“编程蛋白质”的时代。
在这一领域,最具代表性的团队之一就是 David Baker 领导的华盛顿大学蛋白质设计研究所(IPD)。这支团队在过去十多年里不断突破,先后开发了 Rosetta、AlphaFold 相关工具,最近更是把深度学习和生成模型引入蛋白质设计。
而我今天要和大家分享的,是基于 Baker 组博士生 黄博伟(Buwei Huang) 的博士论文的引入部分,做了一些我自己的理解,可以方便大家理解蛋白质设计发展的脉络。

在接下来的文章里,我会带你一起走进这篇论文,总结两大核心内容:
蛋白质结合物的概念与设计思路
三代计算设计 pipeline 的演进
1. 蛋白质结合物 Minibinders 的概念
1.1 什么是 minibinders?
简单来说,minibinders 是 人工设计的小型蛋白质,通常只有 50–70 个氨基酸,结构紧凑,功能单一。它们的主要任务是:
高特异性、高亲和力地结合目标蛋白上的某个特定位点(epitope)。
如果说传统的抗体像一把大雨伞,能牢牢罩住一个靶点;那么 minibinder 更像是一把小巧的折叠伞,精准地卡在你想要的位置上。
1.2 为什么要做小蛋白?抗体不够好吗?
抗体当然强大,它们是目前最重要的生物药物类别(占了全球生物药物市场的大头)。但抗体也有不少“短板”:
体积太大:动辄上百千道尔顿,很多组织(比如脑部)难以进入。
生产复杂:需要哺乳动物细胞培养,成本高。
稳定性有限:抗体在高温或极端环境下容易变性。
而 minibinders 的优势正好互补:
小巧稳定:几十个氨基酸,热稳定性极佳(很多能耐受 95°C 加热!)。
高特异性:计算机可以精确控制它们结合的氨基酸位点,避免副作用。
生产便宜:像普通蛋白一样,大肠杆菌就能表达出来。
模块化组合:可以像乐高积木一样拼接,做多价、多功能的“定制药物”。
1.3 它们能做什么?
在论文中,黄博伟主要展示了两大方向:
作为拮抗剂:阻断关键蛋白信号通路,比如细胞因子风暴中的 IL-6、IL-1 通路。
作为内吞诱饵(EndoTags):把靶标蛋白“骗进”溶酶体里进行降解,类似一种“自毁标签”。
这两个方向都直击传统药物的痛点:
小分子药物虽然能进入细胞,但很难做到高特异性。
抗体药物虽然特异,但大、贵、不稳定。
minibinders 正好卡在中间 → 小巧 + 高特异性 + 可模块化。
可以说,minibinders 是介于抗体与小分子之间的新一代药物平台。它们不是替代品,而是补充,可以填补现有药物的空白。
2. 蛋白质设计 pipeline 的三代演进
在黄博伟的博士论文中,蛋白质结合物(minibinders)的设计被系统性地分为三代方法学演进。这一演进过程,反映了计算驱动与人工智能驱动方法在蛋白质设计领域的结合与迭代。
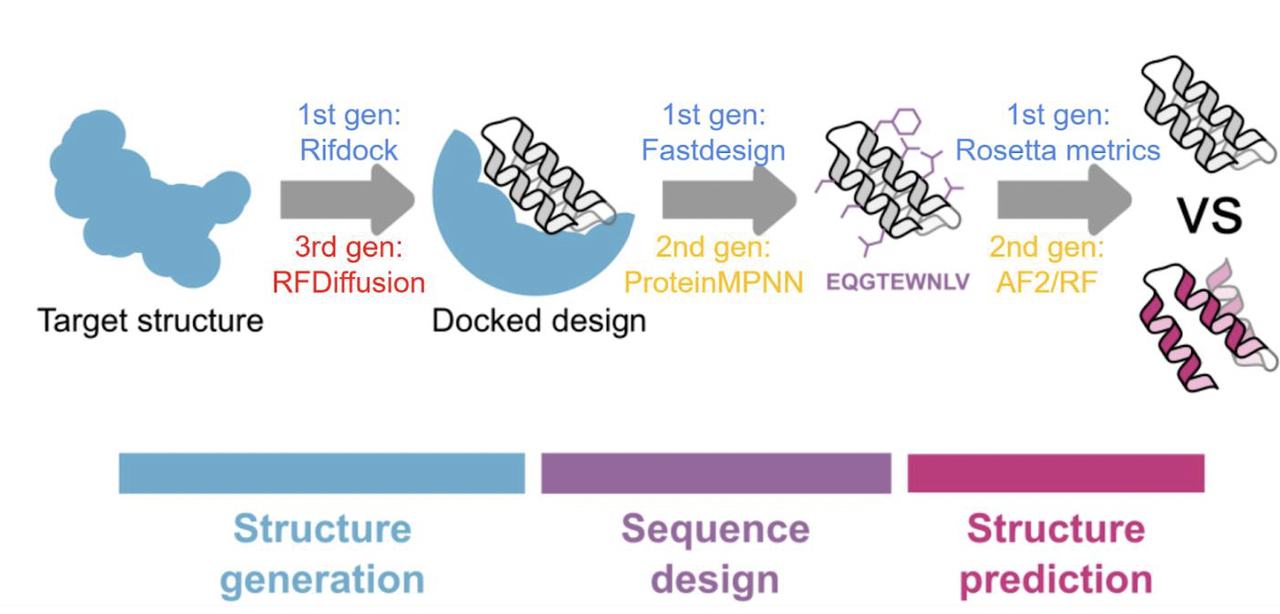
2.1 从经验筛选到基于结构的信息
早期药物开发主要依赖 经验性筛选 或 大规模文库筛选,很难直接锁定目标蛋白上的某个表位 。
一种改进方法是 基于已知结合蛋白的片段嫁接(motif grafting),但这种方式过度依赖现有天然蛋白,导致可采样空间受限,亲和力和适用范围有限 。
2.2 第一代:基于 Rosetta 的结构驱动方法(Cao 2021)
在蛋白质设计的早期,研究者最初的思路是:
先构建一批小型蛋白骨架,然后尝试把它们“拼接”到目标蛋白的结合位点上。
这种方法最具代表性的实现,就是 Baker 团队开发的 Rosetta 设计流程。
2.2.1 骨架库的建立
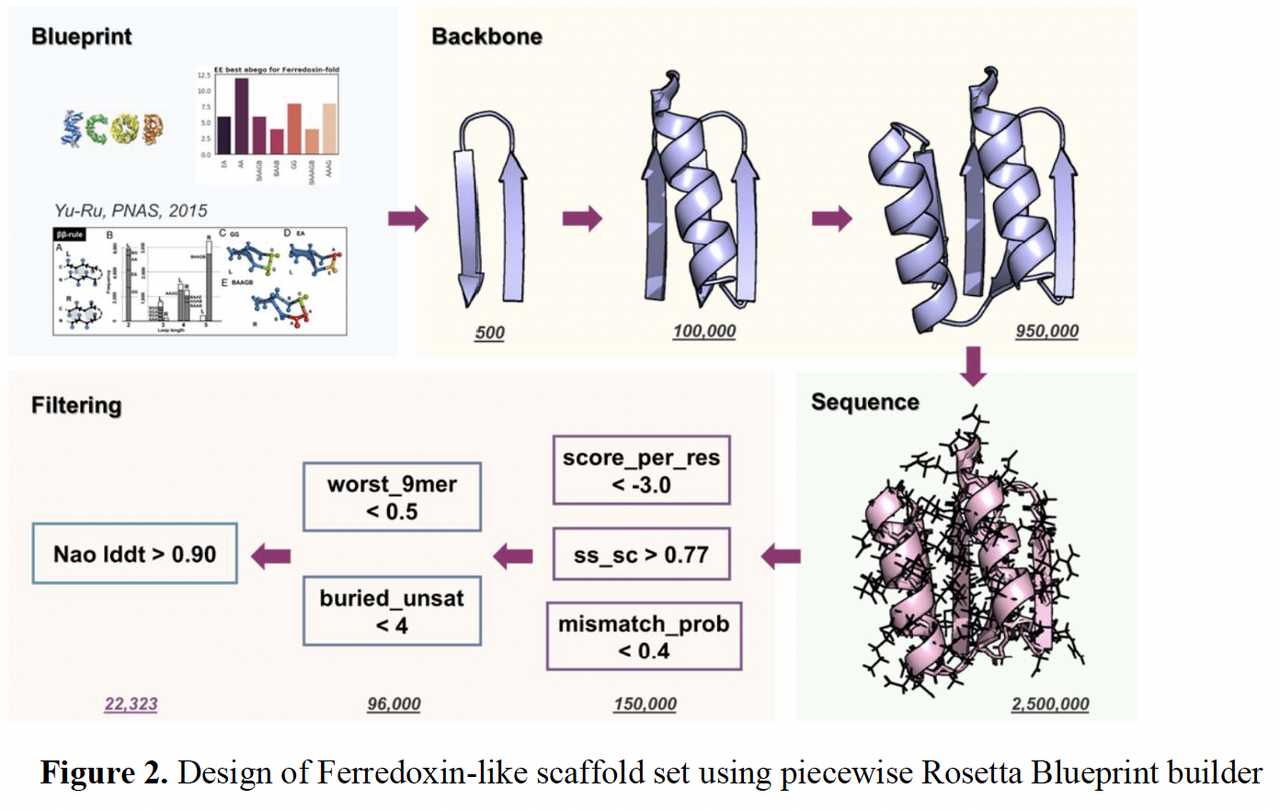
研究者首先需要一个可以尝试的“零件库”。
他们从自然界常见的蛋白质折叠(例如铁氧还蛋白 ferredoxin-like 结构)出发,抽象出常见的二级结构排列模式。
然后通过 Rosetta Blueprint 构建方法,组合不同的 α 螺旋、β 片层和环区,拼装成几十个氨基酸长的小蛋白骨架。
最终生成了一个 多样化的骨架库,每个骨架大约 50–70 个氨基酸,能折叠成稳定的三维结构。
这一步的意义在于:如果没有骨架,就无法承载结合界面。骨架库相当于设计的“备件仓库”。
2.2.2 骨架与目标表位的对接
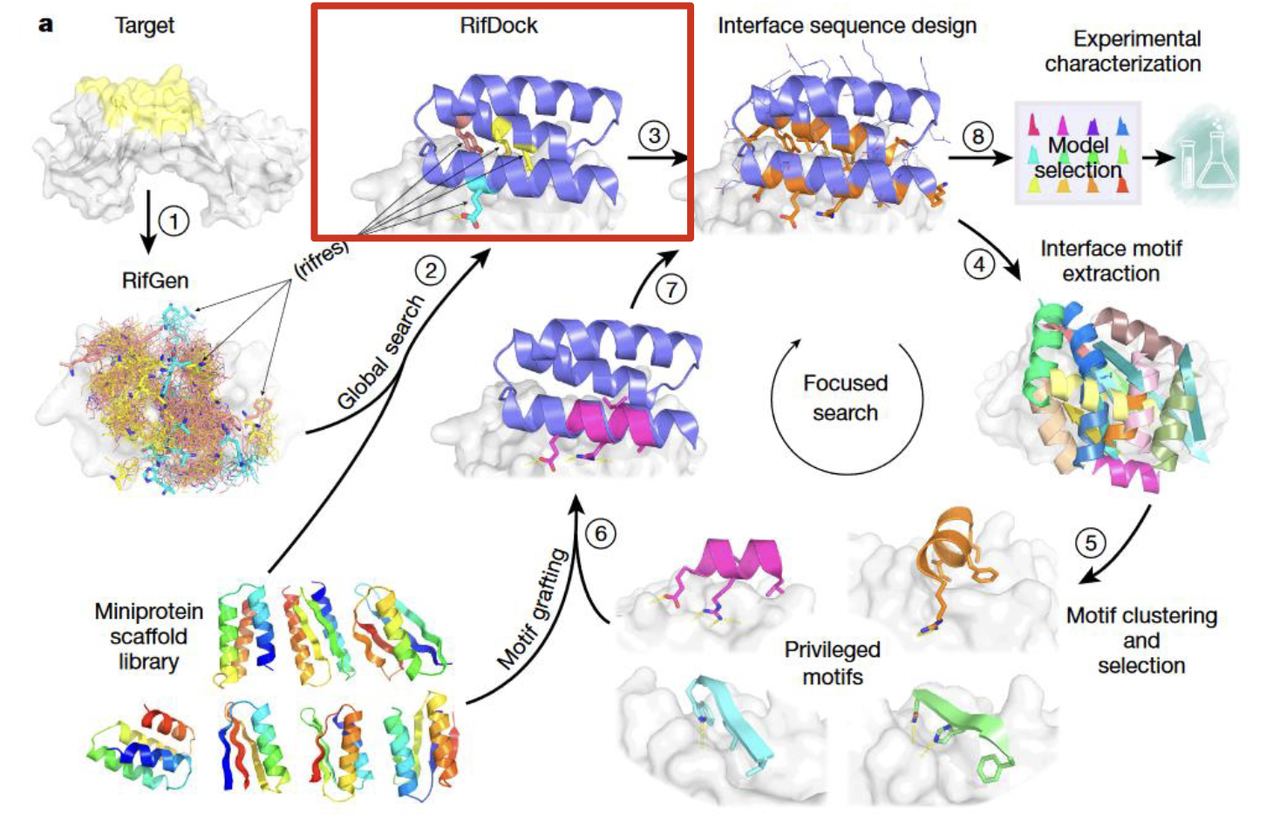
接下来,研究者使用 PatchDock 和 RifDock 这两种分子对接工具,把骨架尝试性地“放置”到目标蛋白的结合表位上。
PatchDock 用于快速找到几何上可行的初始对接模式。
RifDock 则在此基础上进行构象调整和精细化采样,尝试找到最佳的空间互补方式。
这一过程类似“把各种钥匙模具往锁孔里试”,直到找到潜在合适的组合。
2.2.3 序列与能量优化
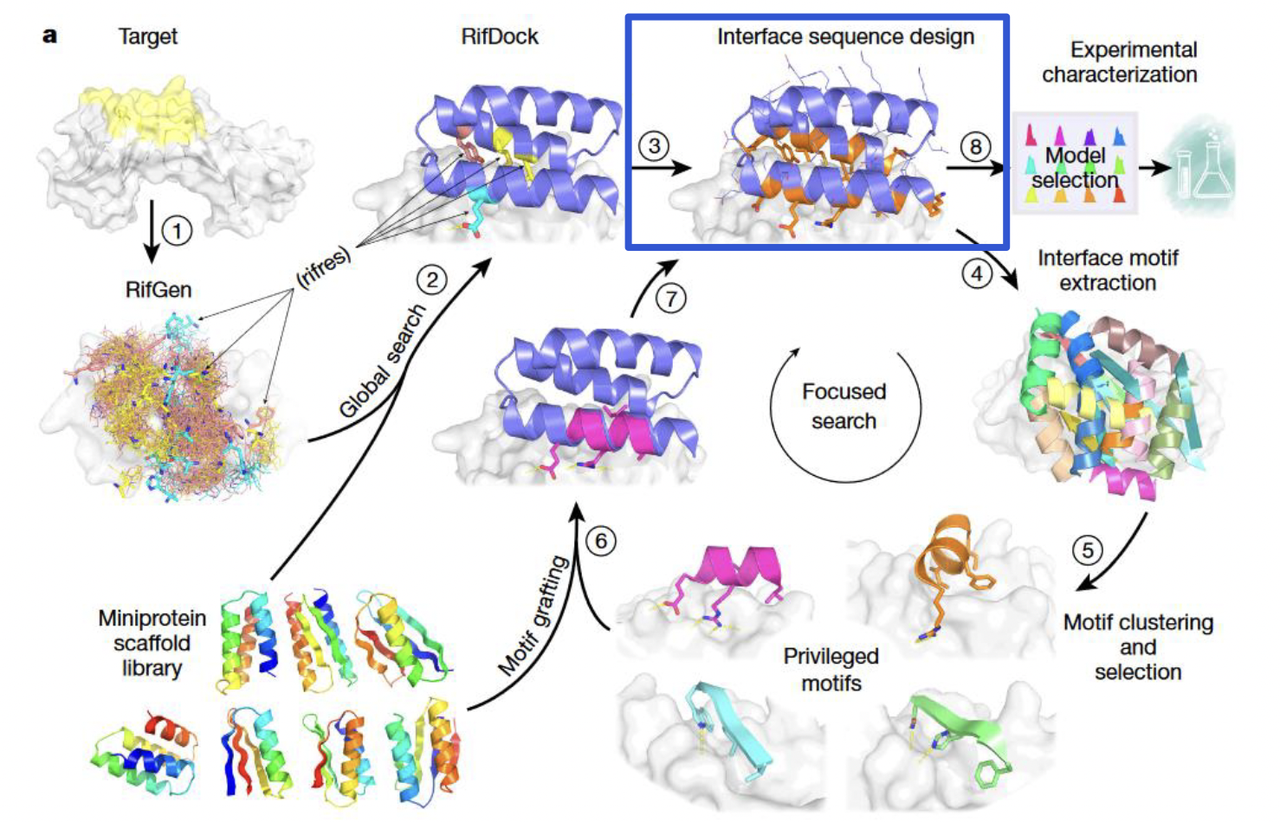
对接后,骨架只是几何上“卡进去”,它的氨基酸序列还需要调整。
研究者使用 Rosetta FastDesign 算法,对骨架的氨基酸序列进行重新设计,优化氢键、疏水相互作用、静电作用等。
同时通过 Rosetta 的能量函数来评估:
结构是否能稳定折叠?
与目标蛋白的界面是否足够紧密、互补?
结合自由能是否足够低?
2.2.4 筛选与实验验证
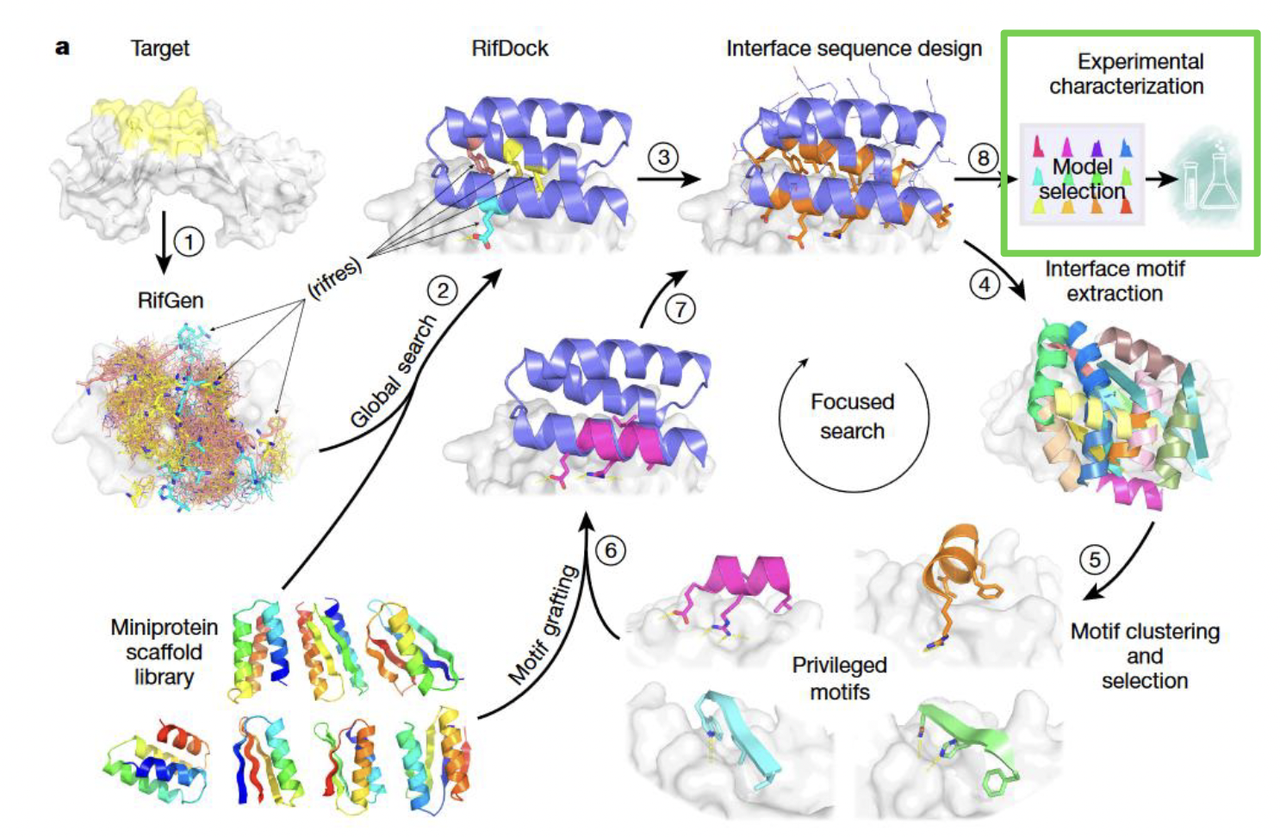
最终,研究者从海量候选设计中,筛选出结合界面面积大、能量评分优的候选小蛋白,送去实验验证。
然而,这种方法面临两个巨大瓶颈:
成功率极低:在数十万甚至百万级别的候选设计中,真正能在实验里稳定折叠并高亲和结合的比例只有 约 0.1%。
计算与实验成本高昂:每一次设计都需要大量对接、优化和筛选,计算资源消耗巨大;实验部分则往往“十万投一中”。
2.2.5 小结
第一代 pipeline 的最大意义在于 证明了从零开始设计结合物是可行的。研究者确实能够利用骨架库和 Rosetta 打分,得到能与目标蛋白结合的小蛋白。但它的低成功率和高开销,使得这种方法 更像是概念验证,而非实用工具。
这一代方法的局限性也为后续的改进指明了方向:
如何让序列设计更高效?如何提升结构预测与筛选的准确性?
2.3 第二代:深度学习辅助的混合方法(Bennet 2023)
在第一代 Rosetta 流程中,虽然证明了 de novo 设计蛋白结合物是可行的,但极低的成功率和高昂的成本严重限制了应用。为此,研究者在第二代 pipeline 中引入了 深度学习工具,大幅提升了效率和准确性。
2.3.1 核心改进一:ProteinMPNN 替代 Rosetta FastDesign
在第一代方法里,序列优化由 Rosetta 完成,需要在巨大的序列空间里不断尝试,计算负担极重。
- ProteinMPNN 的出现改变了这一局面。它是一个基于图神经网络的序列生成模型,能够在已知骨架结构的情况下,快速生成合理的氨基酸序列。
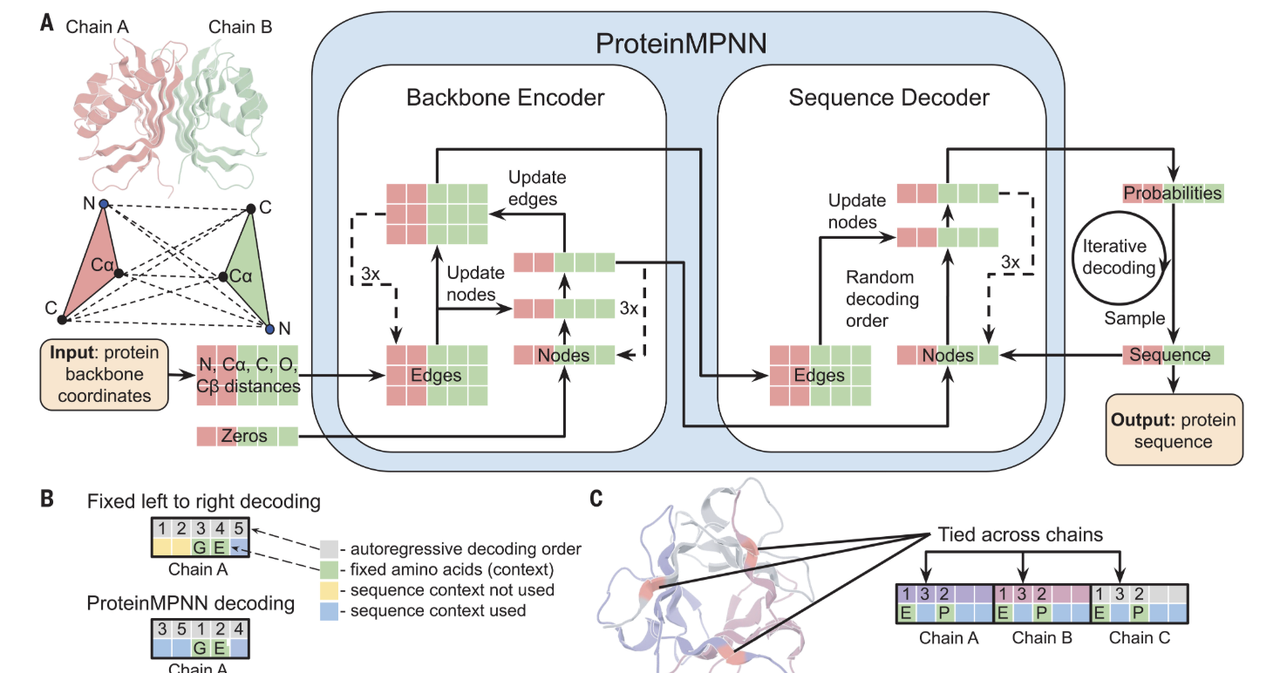
相比 Rosetta FastDesign:
速度更快:数分钟就能完成过去数小时甚至数天的计算。
质量更高:生成的序列折叠稳定性和结合成功率明显优于传统方法。
可以说,ProteinMPNN 把序列设计从“低效搜索”变成了“高效预测”。
2.3.2 核心改进二:AlphaFold2 作为独立打分工具
设计的蛋白质结构,能否真正按照预期折叠?在第一代方法里,这一步依赖 Rosetta 能量函数打分,但其区分度有限。
AlphaFold2 的引入带来了全新的解决方案:
通过预测设计蛋白的折叠结构,验证其是否稳定。
特别是 PAE (Predicted Aligned Error) interaction 指标,可以衡量设计蛋白和目标蛋白结合界面的可靠性。
在实际应用中,PAE 分数成为区分成功设计与失败设计的关键指标,显著提升了筛选的准确性。
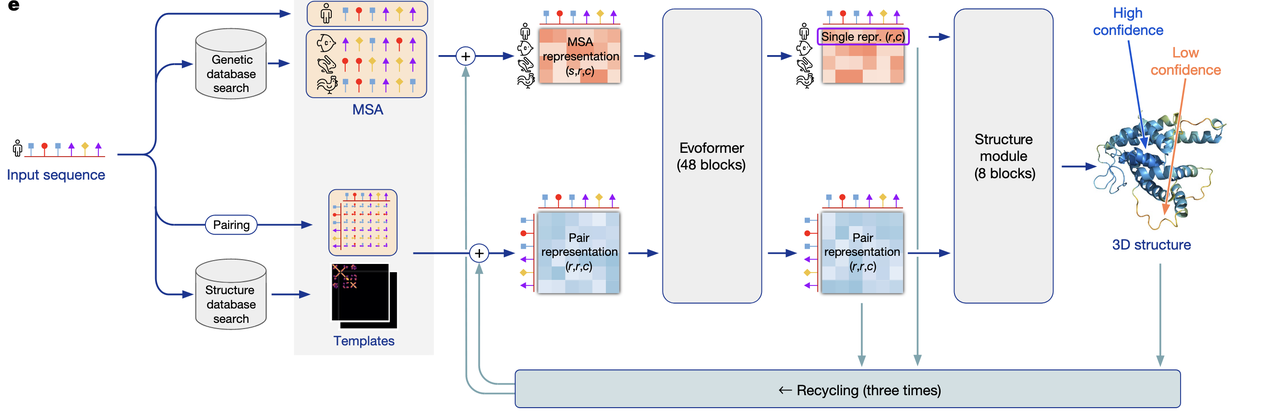
这意味着,研究者不再需要完全依赖实验来淘汰失败设计,计算端就能预判大部分结果。
2.3.3 流程整合
第二代 pipeline 的整体流程可以概括为:
对接阶段:依然使用 Rosetta docking 来探索骨架与表位的初步结合模式。
序列设计:用 ProteinMPNN 快速生成高质量序列。
筛选验证:通过 AlphaFold2 预测折叠和结合界面,利用 PAE 打分筛选最佳候选。
相比第一代“Rosetta 全包”的方式,第二代方法形成了一种 混合 pipeline:
Rosetta 负责几何构象搜索。
深度学习负责序列设计和结构筛选。
2.3.4 成效与意义
成功率显著提升:设计-验证-命中的比例远高于第一代,减少了实验消耗。
计算效率提升数十倍:大幅降低了对计算资源的依赖。
奠定了 AI 融入蛋白质设计的范式:证明深度学习不仅能预测结构,也能直接参与设计环节。
2.3.5 小结
第二代 pipeline 的核心贡献是 将 AI 与传统物理建模相结合,实现了设计流程的降本增效。
它不再是“十万投一”,而是让 de novo 设计真正进入了可以规模化探索的阶段。
不过,这种方法依然有一个限制:仍然依赖预制骨架库。骨架库覆盖不到的表位,依旧很难设计。
这正是第三代 pipeline 要解决的问题。
2.4 第三代:生成模型驱动的 RFdiffusion(2023)
虽然第二代 pipeline 已经利用深度学习极大提升了效率和成功率,但它依然存在一个根本限制:
所有设计仍依赖于预制骨架库。
这意味着如果骨架库里没有合适的结构拓扑,那么某些目标表位仍然无法被覆盖。
为了解决这一问题,研究者引入了全新的思路:直接生成所需的蛋白骨架。这就是第三代 pipeline 的核心——基于扩散模型的 RFdiffusion。
2.4.1 原理:扩散生成模型
扩散模型是一类在图像生成领域取得巨大成功的深度学习方法(如 Stable Diffusion),能够从随机噪声逐步生成符合约束条件的目标结构。
在蛋白质设计中,RFdiffusion 的工作方式是:
输入目标蛋白的结合表位约束信息。
模型从随机结构出发,逐步生成与该表位高度互补的小蛋白骨架。
最终得到一个稳定、可折叠且匹配度高的骨架。
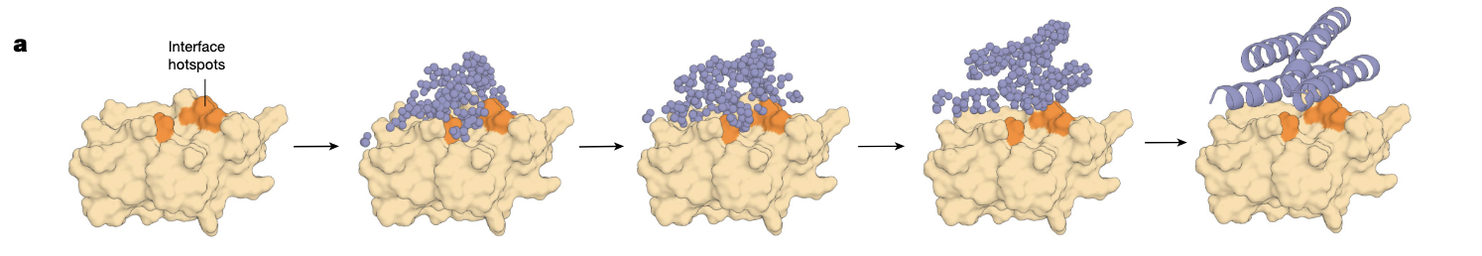
这与之前“从骨架库里挑合适的”完全不同,它是 直接定制化生成骨架。
2.4.2 流程整合
第三代 pipeline 的整体流程:
RFdiffusion:在目标表位约束下直接生成蛋白骨架。
ProteinMPNN:在生成的骨架上快速生成序列。
AlphaFold2:预测折叠与界面结构,用于筛选和验证。
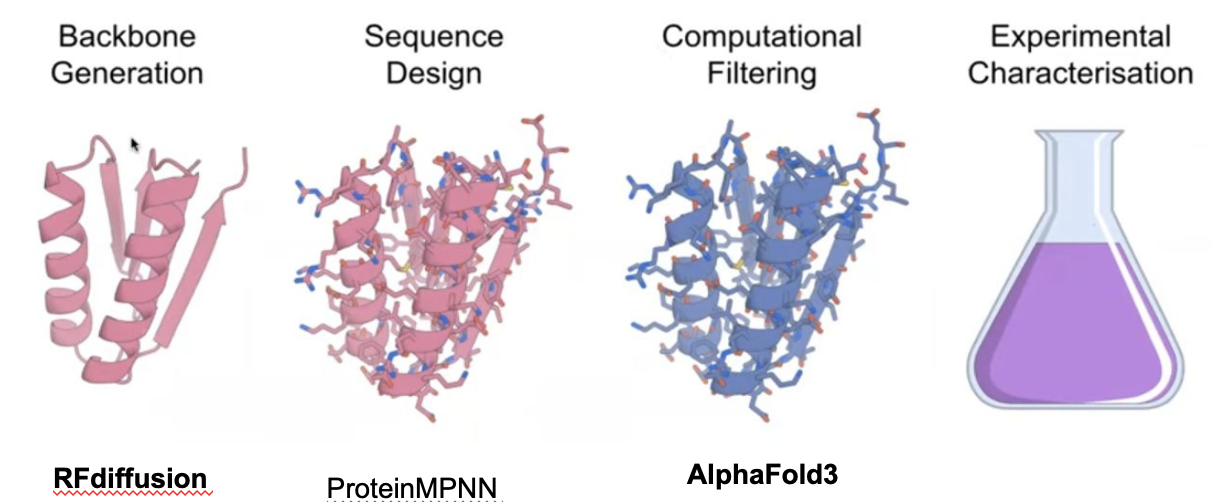
换句话说,RFdiffusion 彻底取代了骨架库和对接步骤,把 pipeline 转变为 表位 → 骨架生成 → 序列设计 → 筛选 的流程。
2.4.3 成果与优势
成功率数量级提升:binder 设计的实验命中率从第一代的 ~0.1%,提升到第三代的 ~10%。这是两个数量级的跨越。
覆盖范围更广:不再受限于骨架库,可以针对几乎任意表位进行设计。
效率与可扩展性:RFdiffusion 能快速生成多样化的骨架,支持大规模并行探索。
2.4.4 意义
第三代 pipeline 的意义不仅仅是提高了成功率,更在于 范式的转变:
从“搜索与优化” → “直接生成”。
从依赖有限骨架 → 实现无限空间的骨架定制。
从尝试性探索 → 走向真正的“按需编程”。
这标志着蛋白质设计进入了一个新的阶段:研究者不再受限于已有结构,而是可以通过生成模型 直接“画”出分子机器的蓝图。
2.4.5 小结
第三代 pipeline(RFdiffusion)实现了质的飞跃,使蛋白质设计摆脱了骨架库的限制,并大幅提升了实验可行性。
这一突破奠定了 可编程蛋白质设计 的基础,也预示着未来的药物开发能够在计算机中实现更大规模、更高精度的分子创造。
3. 总结
这三代 pipeline 的演进体现了蛋白质设计的技术轨迹:
从基于物理规则的穷举搜索(Rosetta),
到深度学习辅助的高效混合方法(ProteinMPNN + AlphaFold2),
再到生成模型主导的按需设计(RFdiffusion)。
随着方法学的进步,蛋白质 binder 的设计成功率与精确性得到数量级提升,这为后续的应用研究(拮抗剂开发、靶向降解等)奠定了坚实基础。
4. 方法学演进总结
通过第一章的梳理,可以看到蛋白质结合物设计在过去几年经历了三个迭代阶段,每一次进步都解决了前一代的瓶颈:
4.1 从骨架库出发的物理建模(第一代)
核心思路:依靠 Rosetta 物理能量函数 + 预制骨架库。
优点:首次证明了 de novo 设计蛋白结合物是可行的。
缺点:
成功率极低(~0.1%),计算与实验开销巨大。
严重依赖骨架库 → 不能覆盖任意表位。
4.2 AI 辅助的混合 pipeline(第二代)
核心改进:
ProteinMPNN → 高效序列设计。
AlphaFold2 → 高精度结构预测与界面打分。
优点:
成功率显著提高,实验命中率增加数十倍。
计算成本大幅下降,设计周期缩短。
缺点:
依旧依赖骨架库。
对新颖拓扑或复杂表位的适应性有限。
4.3 生成模型主导的全新范式(第三代)
核心创新:RFdiffusion 扩散模型 → 在表位约束下直接生成定制化骨架。
优点:
实现“按需生成”,不再受限于骨架库。
成功率提升到 ~10%,相比第一代引入百倍跃升。
意义:从“搜索与优化”彻底转变为“生成与编程”。
4.4 整体脉络总结
早期阶段:依赖天然蛋白改造(motif grafting、筛选)。
第一代:验证了 de novo 设计的可行性,但效率低。
第二代:引入深度学习,大幅提高成功率和效率。
第三代:生成模型实现范式转变,奠定“可编程蛋白质设计”的基础。
4.5 启示
每一代方法的突破,都源于 新工具的引入:
Rosetta → 物理能量函数
ProteinMPNN & AlphaFold2 → 深度学习
RFdiffusion → 生成模型
整个领域的发展趋势非常清晰:
从依赖自然和物理规则 → 过渡到 AI 辅助 → 走向 AI 主导。
这不仅是蛋白质设计的技术迭代,也是 生命科学与人工智能深度融合 的缩影
蓝极说:
回顾这三代蛋白质设计方法的发展,我们看到的不仅仅是算法和技术的更新迭代,更是一个科学范式的转变:从依赖自然、依赖偶然的“寻找”,到可以借助 AI 的力量主动“创造”。第一代方法让我们看到了可能性,第二代方法让这种可能性变得更高效、更可控,而第三代方法则真正把我们带入了“编程蛋白质”的新纪元。
这意味着,未来的药物不再只是自然界里“碰巧存在”的分子,而是可以根据疾病的需求被量身定制的分子机器。蛋白质设计从概念验证走向大规模应用,也许正如计算机从最初的实验室原型走向普及一样,将彻底改变医学与生物技术的格局。
原文链接:https://digital.lib.washington.edu/server/api/core/bitstreams/bd74c9ab-2bd4-44ef-a68f-329f194e3226/content
延伸阅读
本文属于 AI4S文献 栏目。
返回 AI4S文献 → 去公众号阅读完整版 →