英伟达 La-Proteina 开源:首个可扩展到 800 残基的全原子蛋白生成模型
就在昨天, La-Proteina 正式开源了。这是 NVIDIA 团队提出的一种能够在 全原子层面生成蛋白质结构的模型。

相比只做 backbone 的方法,La-Proteina 最大的突破在于:它不仅能生成骨架,还能同时生成 侧链与序列,而且可以扩展到 800 残基的长链蛋白。
借着它开源的契机,我们就来一起看看这篇文章,了解它的核心方法和亮点。

原文链接:https://arxiv.org/abs/2507.09466
Github仓库链接:https://github.com/NVIDIA-Digital-Bio/la-proteina
一、背景介绍
在蛋白质设计的研究里,过去主流的方法往往是把“序列”和“结构”分开处理:
要么先生成一条氨基酸序列,再依赖预测模型折叠成结构;
要么先设计蛋白骨架,再寻找能匹配该骨架的序列。
这种“先后分离”的范式虽然推动了领域的发展,但它在精确控制功能位点(比如活性中心、结合口袋)时往往显得力不从心。原因在于,真正决定功能的往往是侧链的三维构象,而这需要模型能够在全原子层面同时理解并生成。
然而,直接做全原子生成并不容易:
侧链原子数量因残基不同而变化,序列是离散的,坐标是连续的,两者混合在一起会让建模异常复杂。
很多已有方法在这个环节要么生成质量有限,要么无法扩展到大分子。
就在这种背景下,NVIDIA 团队提出了 La-Proteina —— 一种基于“部分潜变量 + 流匹配”的新型生成模型。它既能兼顾 backbone 的显式建模优势,又能高效处理侧链和序列细节,从而首次在 全原子蛋白生成 上实现了强大的可扩展性。
二、核心方法:部分潜变量的巧思
La-Proteina 的关键创新是引入了一种 “部分潜变量(partially latent)”的蛋白表示。它把问题拆成两层:
- 显式建模骨架(α-碳坐标)
Backbone 是所有残基共享的“主干框架”,在蛋白设计中已经有成熟的生成方法。
保持 backbone 在显式空间建模,可以继承现有 backbone 生成模型的优势,同时保证全局几何合理。
潜变量压缩侧链与序列(per-residue latent variables)
每个残基的序列信息和全原子侧链坐标,被编码成一个固定维度的 latent 向量。
这样不论是精简的甘氨酸,还是庞大的色氨酸,最终都被映射成同样维度的潜变量。
这种设计避免了 “残基侧链原子数不一致” 带来的维度不规则问题,把原本的混合离散-连续建模问题转化为统一的连续空间问题。
- 两阶段训练流程(对应 Figure 1)
Encoder:把真实的全原子蛋白编码为 backbone + per-residue latent。
Decoder:接收 backbone 和 latent,重建出完整的全原子结构与序列。
Flow Matching 模型:在 backbone 与 latent 的联合空间进行流匹配训练,从而学会生成新的样本。
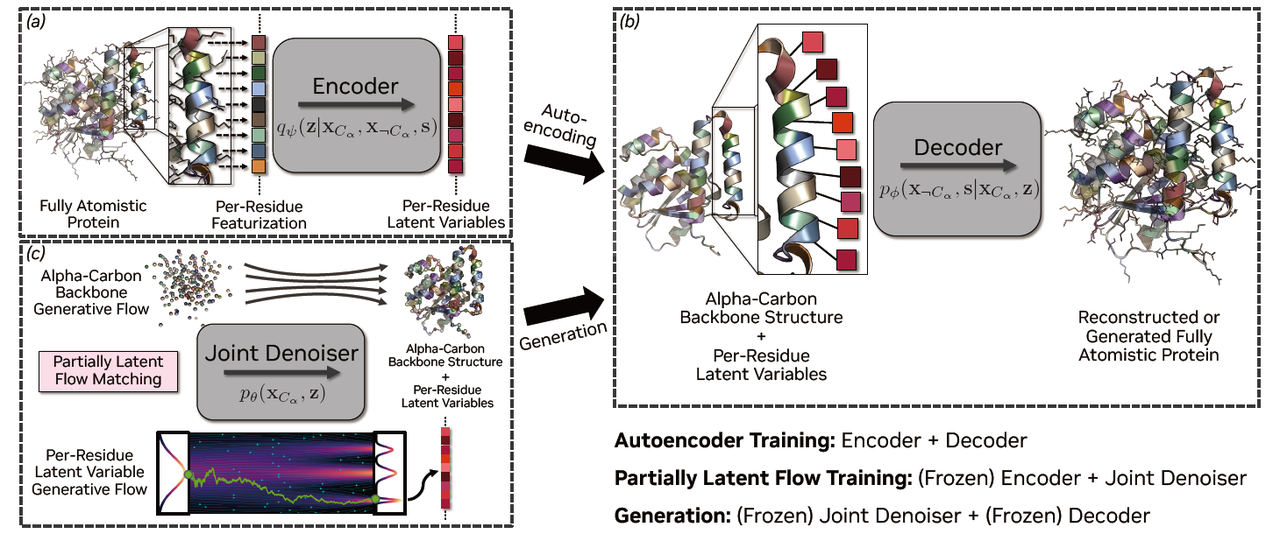
直观理解(结合 Figure 1):
Encoder/Decoder 负责“翻译”蛋白:原子细节被翻译成 latent,再由 latent 翻译回来。
Flow Matching 就像一个生成引擎,它在 backbone+latent 的联合空间里“画出生物合理的轨迹”,最终解码为新的全原子蛋白。
这种混合框架有几个技术亮点:
避免难题:绕开了直接在侧链原子坐标空间建模的复杂性。
灵活可控:骨架和原子细节可以用不同的生成节奏(不同 schedule)采样。
高度可扩展:每个残基 latent 只是额外的通道,不会让模型随残基长度爆炸性增长,从而可以扩展到超长蛋白(后文 Figure 4 会展示 800 残基的生成)。
三、效果展示:从短链到长链
La-Proteina 最直观的成果,就是它能够生成真实可信的全原子蛋白结构,并且在长度上突破了此前模型的极限。
- 短链样本的质量(对应 Figure 2)
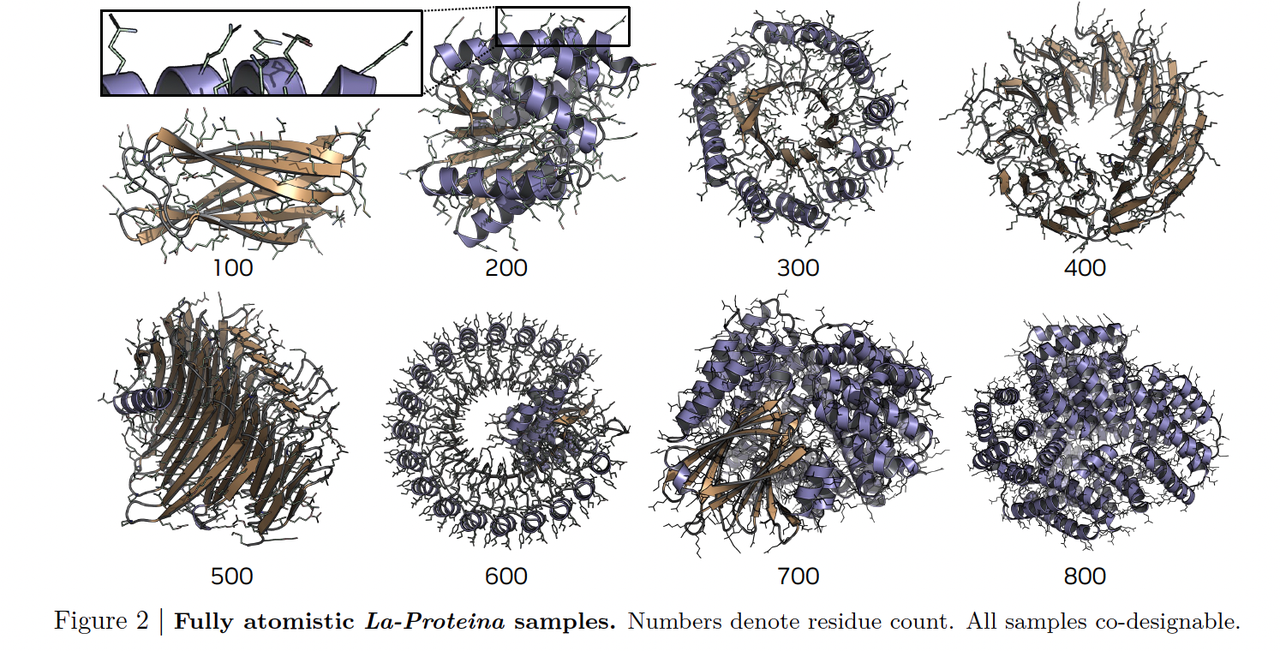
Figure 2 展示了 La-Proteina 直接生成的多个全原子蛋白样本(不同残基数)。
不论是 100 个残基的小型蛋白,还是接近 800 个残基的超长蛋白,生成结果都能保证 backbone 与侧链的协调性。
更重要的是,这些样本不仅外观看起来合理,还通过了 co-designability 测试(即生成的序列能自洽地折叠回生成的结构)。
这说明 La-Proteina 的生成并不是“摆拍”,而是真正具备序列-结构一致性。
- 长链生成的突破(对应 Figure 4)
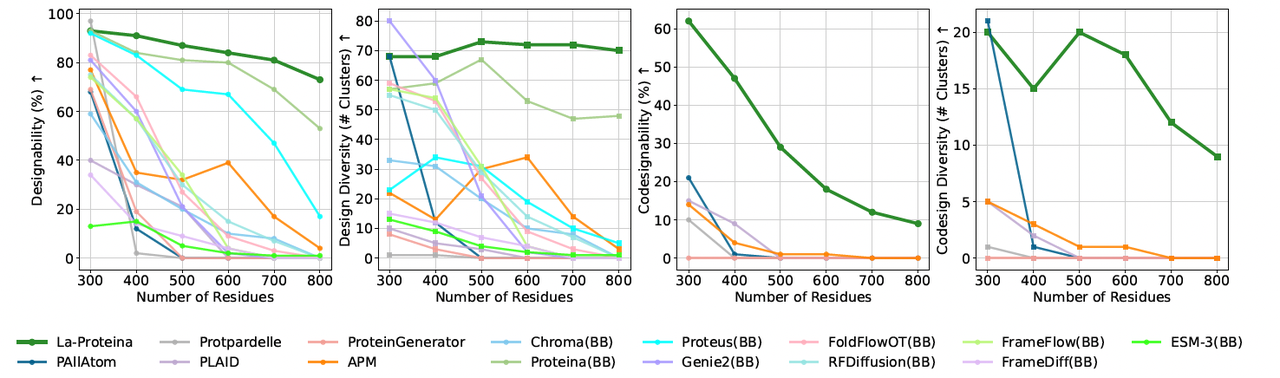
以往的全原子模型在面对长链时普遍会崩溃:
超过 500 个残基时,大多数 baseline 要么无法生成,要么结构严重失真。
例如 P(all-atom) 模型生成一条 500 残基蛋白,就需要 超过 140GB 的显存,几乎不具备可扩展性。
而 La-Proteina 展现出了惊人的稳健性:
它能够稳定生成 800 残基的全原子结构,并保持高 co-designability 和结构多样性。
在 backbone 设计和全原子设计两类指标上,La-Proteina 都显著领先。
相比前一代 Proteina(只做 backbone),La-Proteina 在 backbone 和全原子层面都全面超越。
技术解读
这种跨越式的表现,正是部分潜变量框架带来的:
backbone 的显式建模,确保了大尺度几何的稳定;
per-residue latent 避免了显存爆炸,让模型能承载超大规模数据。
因此,La-Proteina 是第一个真正把 全原子生成 扩展到 长链蛋白 的方法。
四、结构质量与物理合理性
生成蛋白质最大的风险之一,是结构虽然“肉眼可见”合理,但在物理层面可能不稳定、不自然。La-Proteina 在这方面做了系统性的验证。
- MolProbity 分析:整体物理质量(对应 Figure 5)
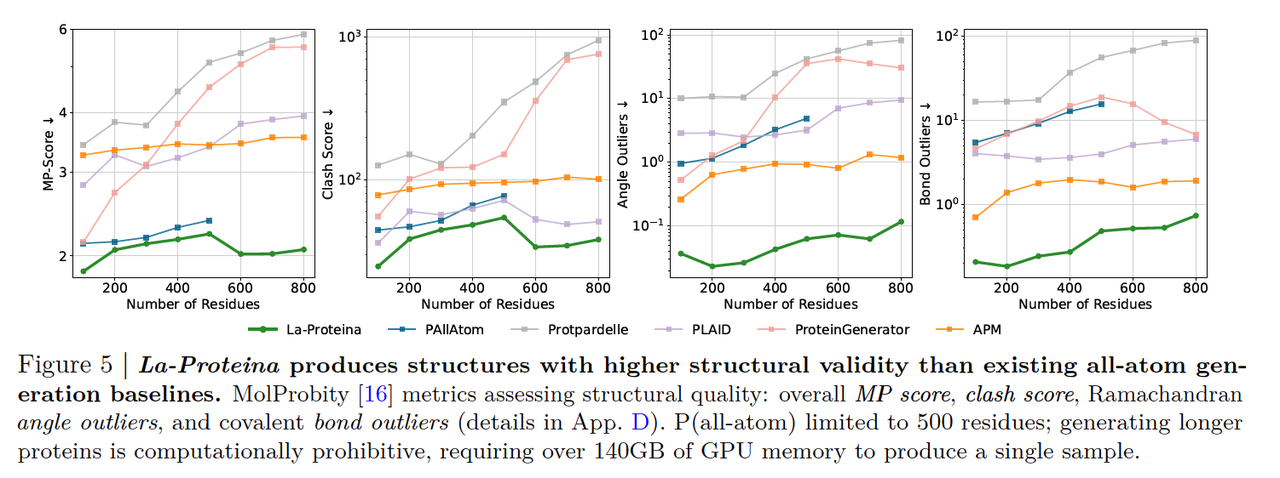
研究团队使用 MolProbity(一个常用的结构质量评估工具)对生成结果进行检查。主要指标包括:
Clash Score:原子间是否存在物理上不可能的重叠。
Ramachandran Outliers:主链二面角是否落在合理区域。
Bond Outliers:共价键长、键角是否合理。
结果显示:
La-Proteina 在所有这些指标上都显著优于现有全原子生成模型。
生成的结构整体物理质量接近 PDB/AFDB 真实蛋白的水准。
换句话说,La-Proteina 的样本不仅是“几何上长得像”,而且是“物理上能站得住”。
- 侧链构象合理性:Rotamer 分布(对应 Figure 6)
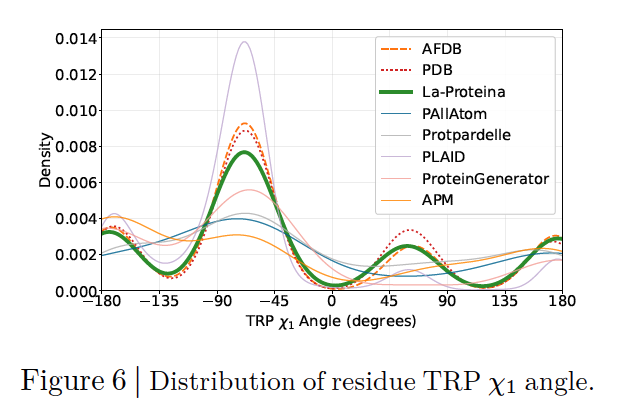
蛋白质的侧链并不是随意摆放的,而是受到空间排斥限制,二面角(χ 角)会聚集在有限的构象状态——rotamers。
Figure 6 以色氨酸(TRP)的 χ1 角为例,展示了 La-Proteina 生成结果的分布。
可以看到,La-Proteina 的 rotamer 分布几乎与 PDB/AFDB 数据重合,能够恢复主要的构象模式和相应频率。
相比之下,其他 baseline 模型要么漏掉了某些模式,要么生成了不合理的角度。
技术解读
这说明 La-Proteina 的全原子生成结果并非“模糊平均”,而是真正捕捉到了蛋白质侧链的 构象能量面 与 生物物理规律。
backbone 的合理性确保了整体框架的稳固;
per-residue latent 的细致表达,使得侧链几何也能自然收敛到合理状态。
五、关键应用:原子级 motif scaffolding
蛋白质设计的一个重要目标,是把特定的功能基序(motif)嵌入到一个合理的整体结构中。这些 motif 往往对应关键功能位点,比如:
酶的活性中心
抗体或配体的结合口袋
金属离子或小分子结合的原子团
过去的大多数生成模型,只能在 backbone 层面做 motif scaffolding(即只考虑 α-碳骨架的空间位置)。但这种粗粒度的方法不足以保证功能位点在原子级别的精确定位。
- La-Proteina 的突破(对应 Figure 3)
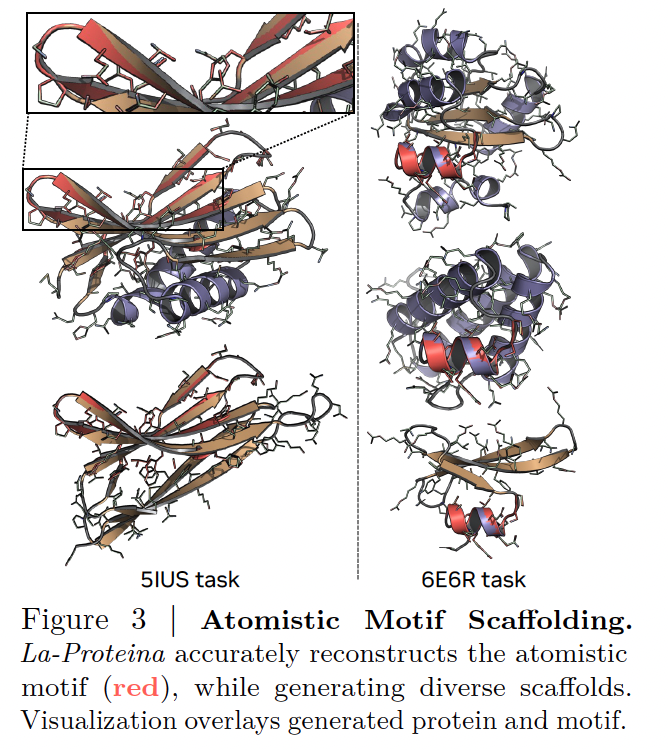
La-Proteina 能够在 全原子层面进行 motif scaffolding:
All-atom scaffolding:输入 motif 的完整 backbone+侧链原子,生成支撑该 motif 的完整蛋白。
Tip-atom scaffolding:只输入 motif 中关键的功能性基团(如末端原子),让模型自动补全 backbone 和侧链。
Figure 3 展示了 La-Proteina 在不同任务上的示例:红色部分是目标 motif,模型生成的 scaffold 能够把它嵌入在一个稳定、多样的蛋白框架中。
- 性能验证(对应 Figure 7)
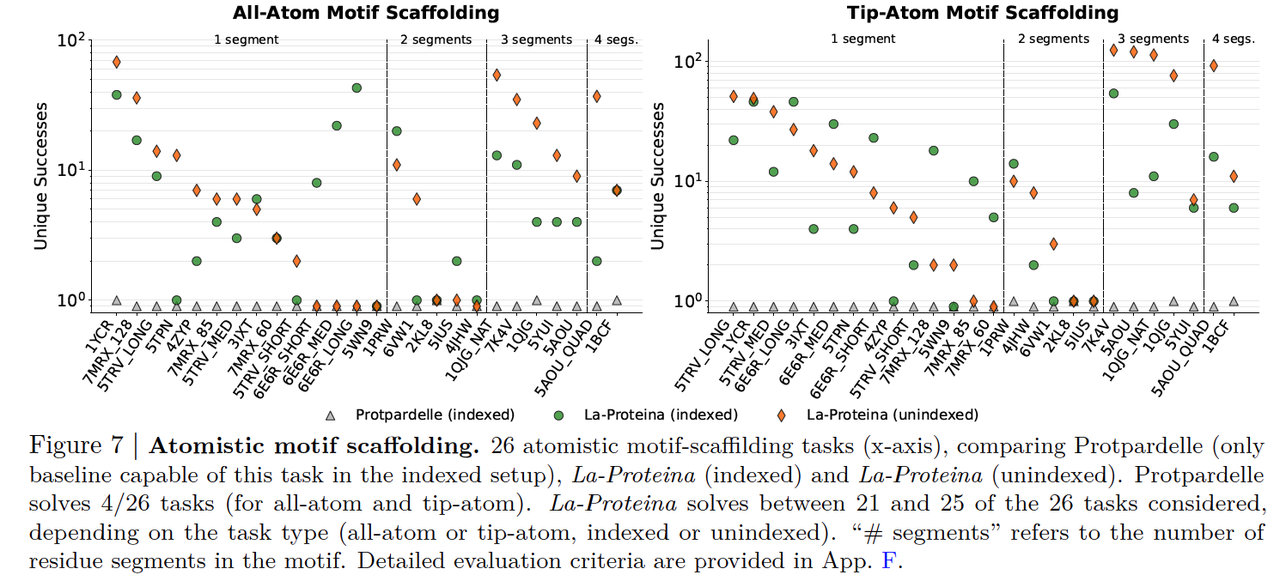
研究团队在 26 个基准任务上测试了 La-Proteina,并与唯一能部分完成该任务的 baseline(Protpardelle)对比:
Protpardelle:仅能在 indexed all-atom scaffolding 下完成少数任务(4/26)。
La-Proteina:在 all-atom 和 tip-atom,以及 indexed 和 unindexed 四种设置下,大部分任务都能成功(21–25/26)。
尤其是 unindexed 情况下,La-Proteina 甚至优于 indexed:
当 motif 残基分散在多个片段时,固定索引会限制结构多样性;
而 unindexed 允许模型自由探索残基位置,反而更容易找到合适的 scaffold。
技术解读
这一结果表明:
La-Proteina 不仅能生成物理合理的蛋白,还能在 功能设计 上实现突破。
原子级 scaffolding 意味着研究者可以直接在生成过程中控制关键功能原子的位置,为 催化酶设计、抗体设计、分子对接 等应用提供强大工具。
六、模型解释性:潜变量空间分析
除了生成能力和应用性能,La-Proteina 还具备一个常被忽视但非常重要的特性:解释性。研究团队通过对潜变量空间的分析,揭示了它的内部结构和可控性。
- 潜变量的“氨基酸聚类”现象(对应 Figure 8 左图)
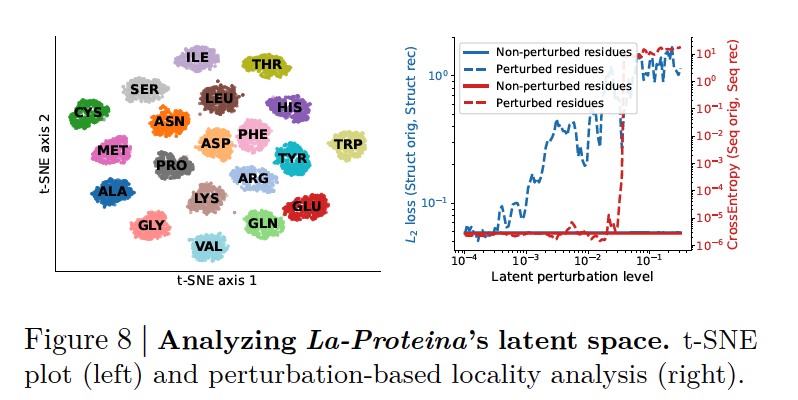
研究者用 t-SNE 将每个残基的潜变量投影到二维空间。
结果显示,不同氨基酸类型在潜变量空间中形成了清晰的簇。
例如:
化学性质相似的残基(如带负电的 Asp/Asn,或芳香族的 Phe/Tyr/Trp)会聚在一起。
这说明模型确实学会了区分并编码氨基酸的物理化学特性,而不仅仅是“记住坐标”。
局部扰动实验:潜变量的“局部性” (对应 Figure 8 右图)
在实验中,研究者对单个残基的 latent 施加扰动,观察解码后的结果。
发现扰动主要只影响该残基的结构和序列,而不会扩散到其他残基。
这表明 每个 per-residue latent 向量主要携带本残基的信息,而不是全局混合。
技术解读
这种“局部化”带来两个好处:
可控性:设计者可以有针对性地修改某个残基,而不必担心影响全局结构。
生物学合理性:氨基酸的局部信息被独立捕捉,同时 backbone 的显式建模又保证了全局一致性。
换句话说,La-Proteina 不仅能生成“像真的”蛋白,还能让研究者对生成过程进行更精细的操控。这是它区别于很多黑箱式生成模型的重要特征。
七、总结与展望
从整体上看,La-Proteina 的贡献可以概括为三个关键词:
全原子、可扩展、可控。
- 全原子层面:
不仅生成 backbone,还能保证侧链构象合理,rotamer 分布与真实蛋白一致。
支持原子级 motif scaffolding,让功能位点的设计进入“亚原子精度”时代。
可扩展性:
部分潜变量的设计,使得模型能稳定扩展到 800 残基级别的超长蛋白生成。
相比需要上百 GB 显存的 baseline,La-Proteina 在计算效率和可用性上实现巨大飞跃。
- 可控性与解释性:
潜变量空间自然地分离出不同氨基酸类型,并保持局部化。
研究者可以针对性地操控某些残基,而不影响整体结构。
局限性
- 数据依赖性强
模型训练用到了超过 4600 万条 AFDB(AlphaFold Database)结构–序列对。
这意味着 La-Proteina 的效果高度依赖大规模高质量数据,普通实验室很难复现同样的规模。
物理真实性仍是近似
虽然生成的结构在 MolProbity、rotamer 分布等指标上接近真实蛋白,但并不是严格的物理模拟。
仍需要后续 分子动力学模拟 或 能量优化 来确认其稳定性和可用性。
- 功能预测缺失
La-Proteina 可以 scaffold 功能 motif,但并没有保证生成的蛋白一定具备预期功能(比如催化活性、结合亲和力)。
在真正应用到药物设计或合成生物学之前,还需要结合功能预测与实验验证。
- 训练和推理成本高
虽然比一些 baseline 节省显存,但主力模型仍有上亿参数(encoder/decoder ~130M,denoiser ~160M)。
大规模训练和长链推理对算力需求依然很高,不适合轻量化场景。
- 解释性仍有限
- 尽管 per-residue latent 展现了良好的局部性和聚类特性,但对于更复杂的长程相互作用,模型的“黑箱性”依然存在。
一句话总结:La-Proteina 把蛋白生成推进到了全原子级别,但它还停留在“结构设计”层面,在“功能设计”与“物理可行性”上仍需要配合其他工具与实验验证。
蓝极点评:
La-Proteina 的提出,标志着蛋白质生成从 backbone 阶段真正迈入了 全原子级别的新纪元。
对科研而言,它能为 酶活性中心设计、结合位点 scaffold、复杂多肽结构探索 提供新工具。
对应用而言,它为药物发现、合成生物学、材料蛋白设计等方向打开了新的可能性。
一句话总结:La-Proteina 让蛋白质设计从“长得像”走向“物理真实、功能精确”,是全原子蛋白生成的重要里程碑。
延伸阅读
本文属于 AI4S文献 栏目。
返回 AI4S文献 → 去公众号阅读完整版 →