结构不是前提,催化才是:RFdiffusion2 重塑金属酶设计
今天给大家介绍一下David Baker课题组这周刚发在Nature上的文章《Computational design of metallohydrolases》。这篇文章宣称:“我们让 AI 直接围着 DFT 给出的催化几何长出一条酶。

我想做的,是把这篇文章的逻辑讲清楚:
为什么旧方法设计不出这样的酶?
RFdiffusion2 为什么能?
ZETA 系列背后到底代表着什么?
下面,我们就顺着文章的图——从噪声到酶,从几何到功能——把故事完整讲一遍。
一、为什么要重新设计金属水解酶?——从一张反应示意图说起(对应 Fig.1a)
这篇文章一开头就给出了一张非常“朴素”的化学反应图:
一个锌离子(Zn²⁺)抓住一分子水,把它变成进攻性极强的 Zn–OH⁻,对准底物 4-methylumbelliferyl phenylacetate(4MU-PA)上的酯键,一刀切下去,生成荧光产物 4-methylumbelliferone 和苯乙酸。
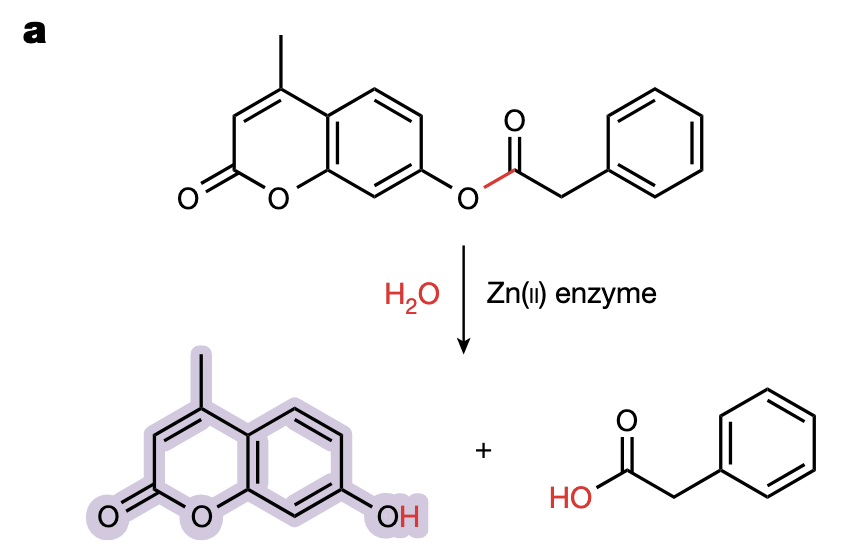
如果你盯着 Fig.1a 多看几秒,会发现作者其实在用这张图暗示三件事:
- 金属水解酶是“高难度反应”的专家
在生物体系里,要在温和条件下切断一个稳定的酯键、酰胺键甚至磷酸酯键,本身就是高难度任务。
单靠水分子自己去攻击,既不够“狠”,也找不准角度。
锌离子一旦登场,就能同时完成三件事:
1)极化底物羰基,
2)活化水分子,
3)在过渡态阶段稳定阴离子中间体。
也正因为这种“多线操作”,天然锌酶(如碳酸酐酶、金属β-内酰胺酶)在催化效率上往往可以达到 10⁴–10⁶ M⁻¹ s⁻¹ 的量级。
现实需求远远超出现有天然酶的“业务范围”
图中选用的 4MU-PA 只是一个实验室用的荧光底物,但作者在正文里很明确地把视角拉向更广的应用场景:
塑料、农药、工业合成中出现的各类“新型”酯类、酰胺类、磷酸酯类污染物;
演化历史上,它们出现得太晚、太“反常”,自然界根本没有足够时间为它们定制高效水解酶。
换句话说,人类造出了大量“没人负责清理”的化学键,而金属水解酶是最有希望接这个活儿的那一类酶。

- 传统 de novo 金属酶设计离“理想状态”还差得很远
文中顺带回顾了前人的工作:
之前的 de novo 金属水解酶,kcat/KM 通常只有几到几十 M⁻¹ s⁻¹;
想要逼近天然酶,几乎都要经过多轮定向进化才能勉强够得着。
也就是说,过去的路线更像是:
先在计算机里凑出一个“能用”的原型 → 再交给实验室用进化慢慢打磨。
而这篇文章想做的,是把路线反过来:
能不能只从“反应需要的关键原子几何”出发,让 AI 一步到位长出一个真正高活性的金属水解酶?

接下来,作者就从这个简单的反应图,一步步走向真正的计算设计框架:
先是用量化计算精确锁定过渡态(Fig.1b),再对比旧一代 RFdiffusion 的局限与 RFdiffusion2 的新思路(Fig.1c),最后用一条“从噪声到酶”的推理轨迹(Fig.1d)把故事铺开。我们下一部分就顺着这组三连图往下讲。
二、旧方法的瓶颈在哪里?——从 Fig.1b–1c 看清“为什么以前设计不出来”
在 Fig.1a 讲清了金属酶催化的核心后,作者紧接着在 Fig.1b–1c 中埋下了文章的第二个主线:
既然知道催化需要什么几何,那么为什么我们还是很难直接设计出高效金属水解酶?
这一部分的三幅图(Fig.1b、Fig.1c、以及 Extended Data Fig.1 的思想)实际上构成一个非常鲜明的“旧方案 vs 新方案”的逻辑对比。
(1)Fig.1b:从量化计算到 theozyme —— 最小催化几何长什么样?
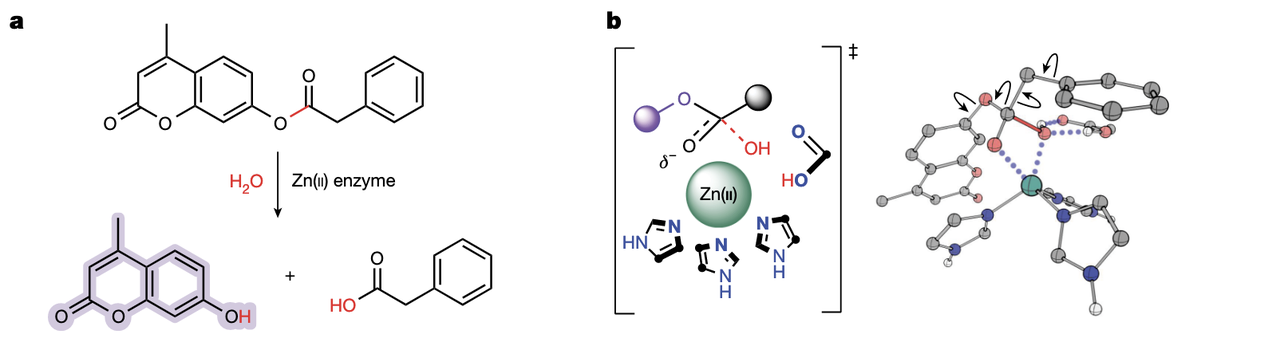
Fig.1b 展示的是作者基于密度泛函理论(DFT)求出的过渡态三维模型:
三个 His 的咪唑环构成 Zn²⁺ 的三齿配位;
Zn–OH⁻ 指向 4MU-PA 的酯键羰基碳,形成典型的四面体过渡态;
还考虑了不同构型的“oxyanion hole”(补偿负电荷的环境)。
为什么这一步重要?
因为过去 de novo 酶设计的一大核心难点是:
你必须提前假设催化残基在哪里、怎么排布。
而 DFT 直接给出了“反应最应该长成的样子”,这相当于把催化问题数学化、几何化——这是后面所有 AI 设计的基础。
(2)Fig.1c:旧版本 RFdiffusion 的根本限制
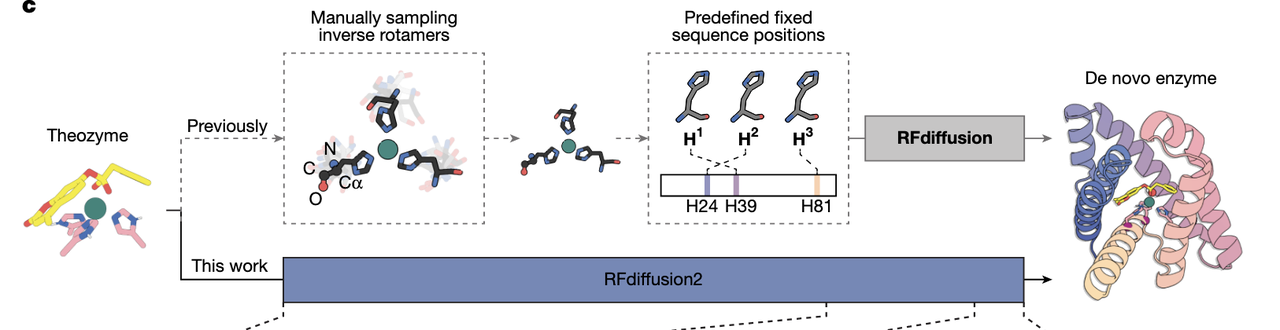
Fig.1c 是整篇文章“问题定义”的关键图。图中的两条路线构成鲜明对照。
旧方法:RFdiffusion(图的上半部分)
旧模型有两个必须“提前指定”的东西:
① 催化残基在蛋白质序列中的具体位置(position)
② 每个催化残基的完整主链构象与侧链转动角(χ1/χ2 等)
这两个需求的问题在图里表现为:
你要把三条 His 放到确切的三个位置(例如某序号的氨基酸);
每条 His 还要指定其侧链旋转情况,以便恰好抓住 Zn²⁺ 并对准过渡态;
甚至还要让骨架延展到合理位置来托住这些 His。
这是什么概念?
就像在没有看到房子的情况下,先决定“客厅灯的螺丝孔在哪里、朝哪边倾斜”,然后再让 AI 去设计整个建筑——荒谬但旧方法只能这样做。
论文计算过:
即便用很粗糙的取样方式,可能组合仍然达到 10¹⁸ 种级别!
这就意味着:
绝大部分输入组合都是不合理的;
模型每次只能看见这一种输入,无法“跨组合推理”;
实际找到催化几何正确的设计 → 靠运气。
(3)新方法:RFdiffusion2(图的下半部分)
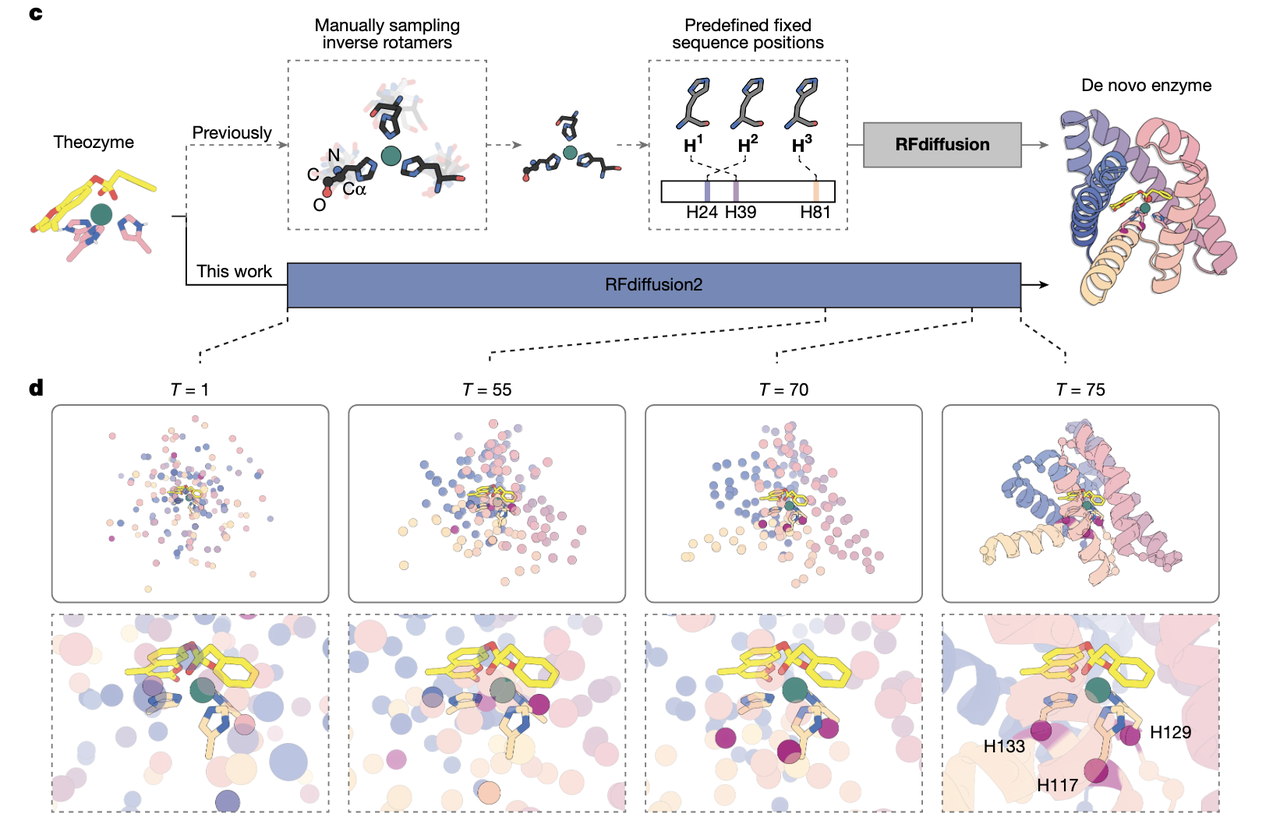
图中蓝色路线展示了完全不同的思路:
输入不再是完整的 His 残基(包含主链与侧链)。
只需要输入关键原子的空间位置:
Zn、三个 His 的 Nε 或 Nδ
过渡态模型的关键原子(C、O 等)。
换句话说,你不再决定 His 在序列中是第 15 位还是第 87 位;
也不再决定 His 的 χ1 是 60° 还是 180°。
模型会在推理中自己决定:
哪个 Cα 应该移动到这些功能团附近;
哪段骨架可以延伸成正确的折叠支架;
哪些序列更能稳定该结构。
三、AI 是怎样从一团随机噪声里长出一条酶的?——读懂 Fig.1d 的诞生轨迹
如果说 Fig.1b–1c 解决的是“为什么以前设计不出来”,那么 Fig.1d 则回答了一个更迷人的问题:
当 RFdiffusion2 真正开始设计时,它眼中的世界是什么样?
更具体一点:
一个酶,是如何从一团完全没有意义的“噪声”中长成现在我们看到的折叠蛋白?
这一幕非常像观看一个加速播放的宇宙诞生纪录片,只不过这里不是星云塌缩成星球,而是一条从未存在过的蛋白质,被“催化几何”牵引着一点点成形。
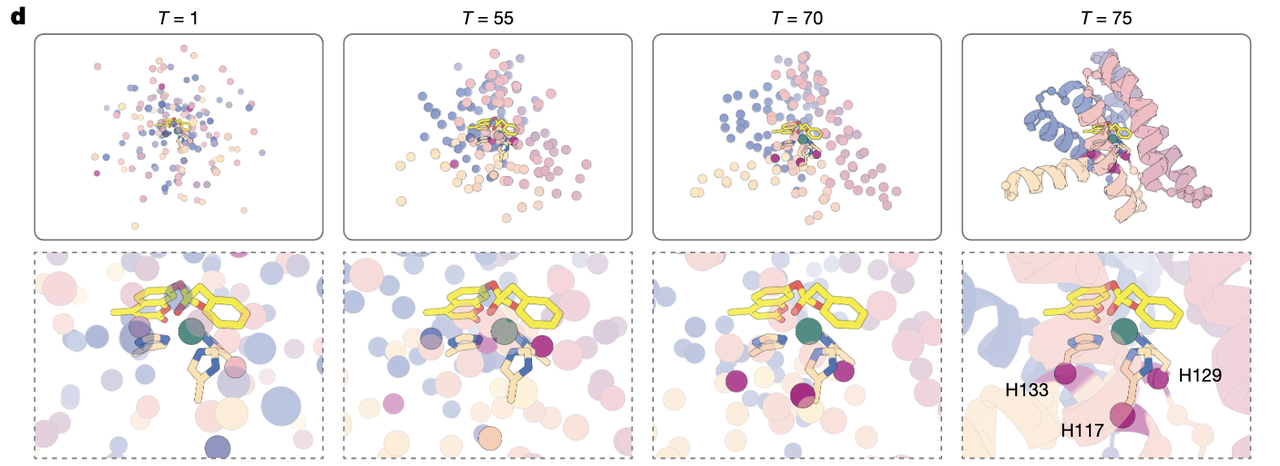
(1)起点:所有 Cα 都是“自由粒子”,它们不知道自己是谁
Fig.1d 的最左侧,是一堆毫无结构的透明点云。这些点就是未来蛋白质的 Cα 原子,每一个都悬浮在空间中,没有连接关系,没有折叠逻辑——
它们一开始甚至不知道它们要成为“第 57 位亮氨酸”还是“第 13 位丝氨酸”。
只有一件事是确定的:
那团来自 DFT 的催化原子(His 的配位原子、Zn²⁺、过渡态结构)被牢牢固定在中心。
这一团结构就像“世界的种子”,它不会动,它等待着周围的混乱逐渐自发变得有序。
(2)中段:结构开始“围着催化核心生长”
随着时间步推进,噪声不再是噪声——
点云开始收缩、聚类、拉出片段,逐渐形成具有方向性的骨架雏形。
这时你会注意到一个非常有趣的现象:
固定在中央的那三个 His 的侧链功能团先出现;
但它们对应的 Cα(也就是整条氨基酸所在的位置)暂时并未就位。
这是整个过程最“魔幻”的时刻:
模型知道这些功能团必须在正确位置,但它还不知道身体应该如何靠过去。
于是你会在中间帧看到一个极不自然但极富启示性的场景:
三个 His 的侧链像“钉子”一样钉在空间里;
但支撑这些钉子的“木板”(蛋白主链)还在远处游荡。
人类设计者绝不可能想象这种过渡态,但 AI 却天然地以这样的方式工作。
(3)收敛:骨架主动“贴上去”,完成连接
当推理进行到后段时,一个神奇的画面出现了:
那些原本在空间另一侧游荡的 Cα 逐渐向催化基团靠拢,
像是被某种力牵引着——那种力来自模型内部对“可折叠蛋白质”的统计认知。
而当某个 Cα 移动到恰到好处的位置时:
它的侧链方向被调整;
结构自动判断:“哦,你应该是承担配位任务的 Histidine。”
于是它与先前固定的 His 功能团自动连接起来。
这种“先有功能团,再长出残基位置”的方式,与我们人类常用的从序列出发建模的思路截然不同:
在人类的世界,是氨基酸决定功能。
在 AI 的世界,是功能决定氨基酸。
这也是 Fig.1d 最值得深玩的一层含义。
(4)终态:蛋白质围绕催化几何“长成一个整体”
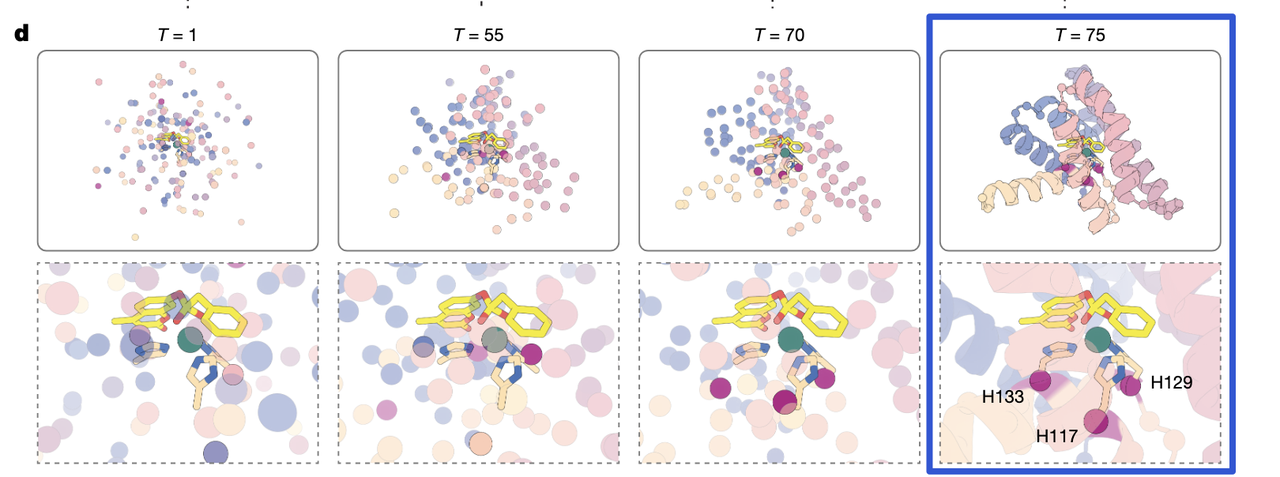
在 Fig.1d 最右侧,你看到的已经是一条颇具自然折叠特征的蛋白结构。
能观察到两个关键点:
催化核心仍然纹丝不动地保持着 DFT 的精确几何;
整个蛋白像是从外向内塌缩,最终形成包裹催化中心的稳定折叠。
其中最值得注意的一点是:
催化 His 的序列位置不是预先写死的,而是在推理过程中“被模型选出来的”。
这意味着 RFdiffusion2 不仅在生成结构,而且在生成“结构与功能的匹配”。
(5)为什么 Fig.1d 的过程如此重要?
因为这段轨迹展示的,是过去二十年酶设计中最难实现的一件事:
让功能先于结构,让结构为功能让路。
传统方法必须先设定结构位置,再塞催化残基进去;
RFdiffusion2 则让催化需求成为整个结构生成的源头——
就像在蛋白宇宙的中心点燃了一颗星核,其周围物质开始按照物理规律与几何约束自然聚集,最终形成一颗前所未有的“催化星体”。
四、第一轮设计:当计算机把一百条候选交到实验室,故事才刚刚开始(Fig.2)
如果说 Fig.1 是“酶如何在 AI 的脑海里生长出来”,那么 Fig.2 展示的,就是这些在数据空间中出生的结构,第一次接受现实世界的考验。
你可以把 Fig.2 想象成一段选择性的叙事蒙太奇:
从电脑里涌出的海量结构,经过层层筛选,最终只剩下少数,等待那盏紫外灯亮起——发光就表示“它是真的动起来了”。
(1)从 5120 条虚拟骨架,到实验室里的 96 条真实蛋白
RFdiffusion2 的设计是一种大规模生成:
5120 条不同的骨架,
每条再经过 ProteinMPNN 生成序列,
再经过 AlphaFold2 预测是否能折回到设计结构,
再用 LigandMPNN 和 Rosetta 对活性位点微调。
这一连串步骤像是给每个候选做了一遍“虚拟模拟训练”:
他们先要在计算世界里证明自己可能折叠、可能稳定、可能抓住 Zn、可能进行催化,才能拿到进入现实世界的“签证”。
最终,96 个设计获得签证,进入湿实验环节。
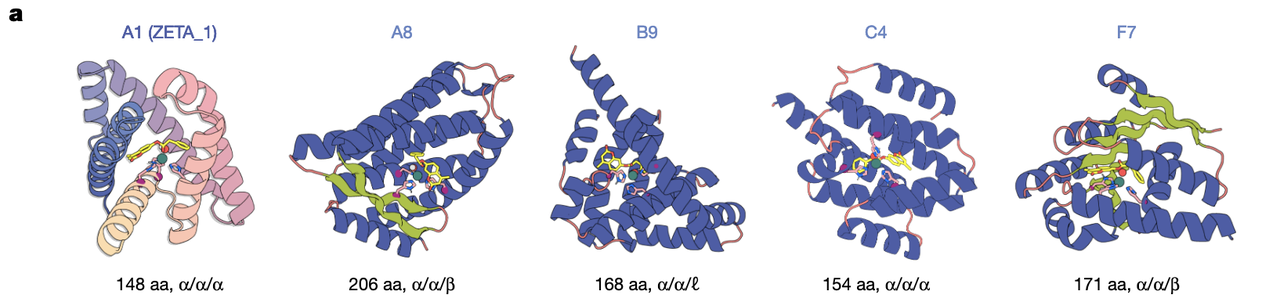
你能从 Fig.2a 看到它们的“身份证照”——
不同长度、不同折叠方式、不同构型,就像是 96 个完全不同风格的陌生人站成一排,每一个都声称:“我可以做金属水解酶。”
(2)实验的第一道门槛:能不能表达?能不能溶?
蛋白质天生挑剔,有些序列折不起来,有些折错了,有些干脆变成包涵体。
Fig.2 没有直接展示这一部分,但正文里给了一个关键数字:
86/96 可以溶性表达。
这是一个令人惊讶的高数字。
在传统 de novo 设计中,溶性表达往往是第一个大坎,能过一半就算运气不错。
而这里 96 个当中有 86 个都“能在细菌里做成蛋白”,说明 RFdiffusion2 生成的结构不仅合理,而且非常容易折叠——这也是一个侧写:AI 在结构生成上的能力已经成熟得远超以往模型。
(3)第二道门槛:能不能动?能不能催化?(Fig.2b)
接下来是 Fig.2b——96 个蛋白各自被加入同一底物 4MU-PA 中,实验人员盯着荧光曲线,等待某个曲线能够真正“亮起来”。
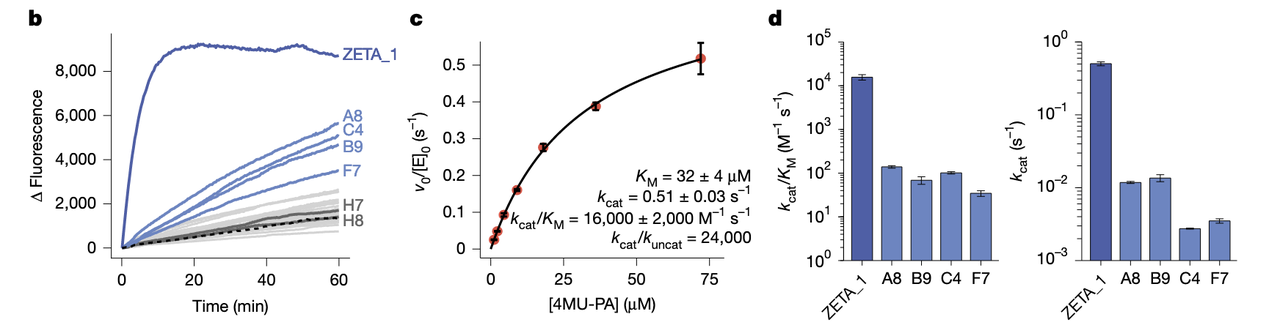
大部分曲线都平得像一条线,但有五条突然崛起:
A1、A8、B9、C4、F7。
图中上升的那一小段斜率,就是计算结构第一次在现实世界里表现出“我是活的”的信号。
这是本文第一次真正意义上的成功时刻:
AI 随机生成的结构中,有 5 条真的会催化反应。
在过去十几年的酶设计中,哪怕你花好几个月人手优化、反复计算、连做几十个变体,也不一定能得到 5 条真正有活性的。
而这次,系统性地第一次尝试,就得到了 5 条。
(4)ZETA_1 的出现:图 2c–2d 讲述的是“一个完全陌生的序列,却拥有天然酶级别的速度”
五条 hits 中最耀眼的是 A1,也就是后来命名为 ZETA_1 的设计。
Fig.2c–2d 展示了它的动力学曲线:
kcat/KM ≈ 16,000 M⁻¹ s⁻¹
kcat ≈ 0.5 s⁻¹
KM ≈ 32 μM
如果你把这些数字放在背景里看,会更震撼:
这是一个完全不存在于自然界的蛋白,序列是 AI 编出来的,结构从噪声中长出来的,却拥有天然锌酶 1–10% 的效率。
在传统 de novo 设计中,第一代原型酶通常是“慢如乌龟”的。能动已经是奇迹,让它动得快更是奢望。往往需要经过十轮八轮的定向进化才能提升一个数量级。
而 ZETA_1 一出生就是高活性的。
这不是偶然,而是 RFdiffusion2 的设计逻辑第一次在实验室被证实为有效。
(5)为什么 ZETA_1 强?Fig.2e–2h 给出第一条答案:它的活性位点几乎“不摇晃”
PLACER 的结果(Fig.2e–2h)非常关键——
它告诉我们:
ZETA_1 的底物在活性位点的位置几乎不动;
催化残基的位置也非常固定;
相比之下,失活设计 H7、H8 的底物在口袋里乱跑,残基也不定位。
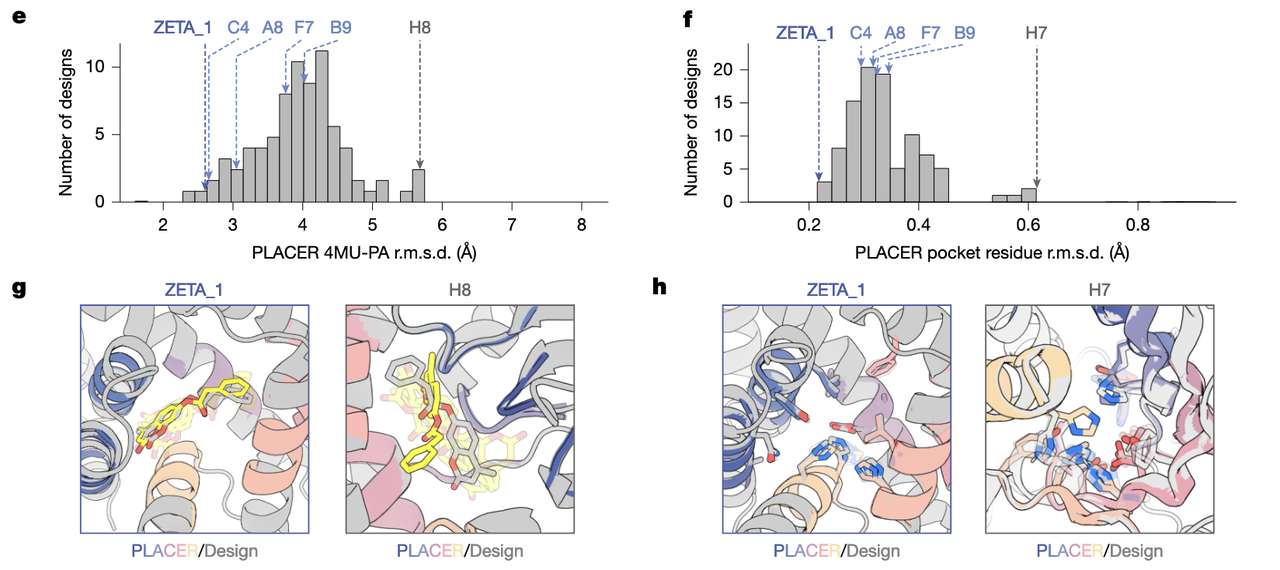
这意味着:
真正的高活性来自“预组织化”——
活性位点的原子已经提前站到正确的位置上,不需要靠诱导契合或大幅构象变化。
ZETA_1 之所以快,是因为它没有“犹豫时间”,底物一来就正位,水分子一来就攻击。
PLACER 的这一段图,是 AI 设计方法学中最重要的证据之一:
它证明了 RFdiffusion2 不是在“随机生成幸运结构”,而是在生成真正具有催化几何的蛋白质。
Fig.2 对整篇文章来说,是一个断点式的时刻:
理论几何(Fig.1)已经被实现;
噪声生成的结构(Fig.1d)在现实中折叠良好;
其中一个结构直接达到天然酶量级的活性(ZETA_1);
活性来源可以由独立方法(PLACER)解释;
并且这些结构与自然界完全不同(Fig.2a)。
换句话说:
酶设计史上第一次,计算机从零开始设计出“能用、好用、还稳定”的金属水解酶,而且是不经优化直接能用的。
这一成就为第二轮设计铺平了道路,也为 Fig.3–5 的深入解析奠定基础。
五、ZETA_1 的解剖:一条从噪声中诞生的酶,为什么一上来就这么强?(Fig.3)
第一轮实验中,五条 hits 像五个“新人演员”站在镁光灯下,而 ZETA_1(A1)就是那个一下子抓住所有人视线的人。
Fig.3 就像对这位“主角”进行的全方位镜头扫描:
它的脸、它的骨骼、它的动作逻辑、它的武器、它的弱点、它对金属离子的依赖,它在高温下的表现,它的结构是不是如 AI 所想——这些问题都在这里一一得到回答。
下面我们就按图的视觉顺序,把 ZETA_1 的故事讲清楚。
(1)Fig.3a:ZETA_1 的“脸”——一个为催化而生的口袋
Fig.3a 左侧是全结构,中间是活性位点,右侧是口袋的表面形状。
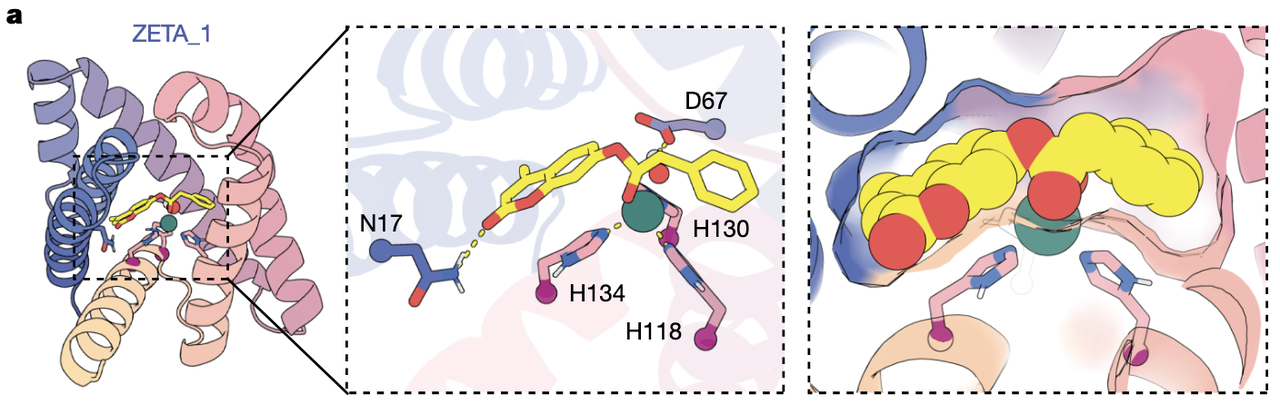
这张图传达了一个非常关键的信息:
ZETA_1 并不是“刚好能催化”,而是显然专门“为催化而折”。
你会注意到三个 His 围着 Zn²⁺ 排列得非常干净,
Asp 位置恰好能充当潜在 general base,Asn 则用一个温柔但关键的氢键抓住底物环系,整个 pocket 干燥、紧凑、疏水、形状互补。
如果你把 Fig.3a 和 Fig.1b 的 theozyme 对比,会发现:
过渡态需要的几何关系,ZETA_1 完整保留了;
甚至连底物进入口袋的角度,都与 DFT 模型高度一致。
这说明 RFdiffusion2 不是凭运气,而是真正学会了“围绕催化原子长出合适几何”。
(2)Fig.3b–d:结构稳不稳?能不能折起来?
这几张图其实回答一个看似简单但极其关键的问题:
ZETA_1 是一条真正能在现实世界折叠的蛋白吗?
● Fig.3b:SEC(凝胶过滤)
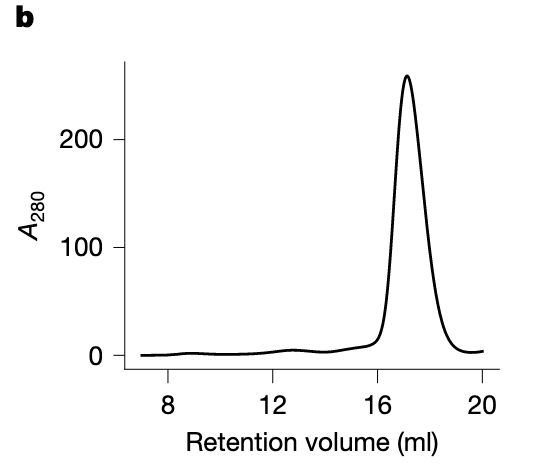
一个漂亮的、清清楚楚的单峰。这意味着 ZETA_1 是单体、均一、没有聚集。
很多 de novo 设计最容易死在这里——设计得好看却不折叠。ZETA_1 显然没有这个问题。
● Fig.3c–d:CD(圆二色)
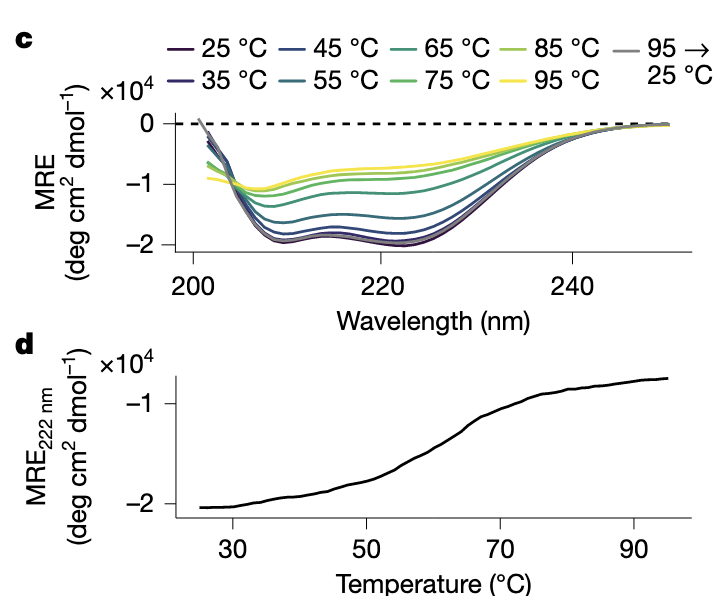
如果你把这两张图连在脑中回放,你会看到:
ZETA_1 随温度上升从 25°C 加热到 95°C;
光谱逐渐改变,但主结构元素仍然存在;
冷却后又基本回到原始状态。
这意味着 ZETA_1 不仅折叠良好,还具备一定可逆的热稳定性。
换句话说,它不只是“能折叠”,而是“折得非常稳”。
(3)Fig.3e:一个关键数字——1000 次以上的 turnover
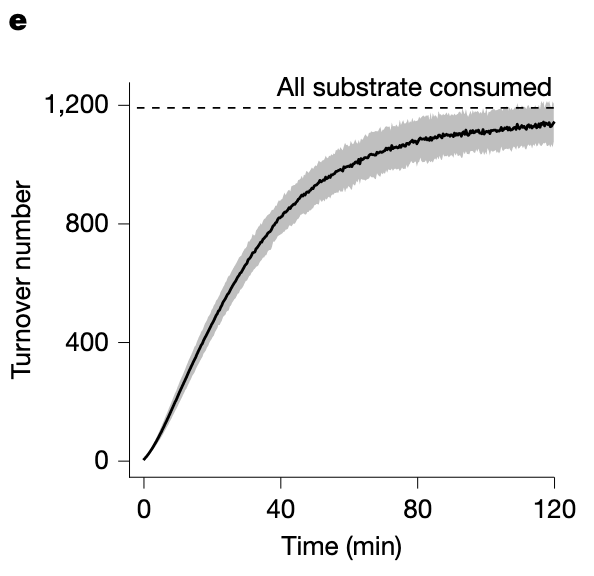
如果 ZETA_1 只是速度快,那叫“瞬时能力强”;
但 Fig.3e 告诉我们:
它可以连续催化超过 1000 次,不明显衰减。
这意味着三件大事:
活性位点没有塌陷;
结构不会被底物或产物破坏;
金属不会轻易流失。
对一个第一次见光的 de novo 酶来说,这种稳定度接近惊人。
(4)Fig.3f–g:ZETA_1 的生命线——Zn(II)
● Fig.3f:拿掉 Zn,它立刻死;加回 Zn,它立刻活。

没有比这更优雅的实验了——
它直接告诉你:
ZETA_1 的催化机理确实是 Zn²⁺-OH 的亲核攻击,而不是一些“假阳性反应”。
● Fig.3g:Zn 结合常数(KD)
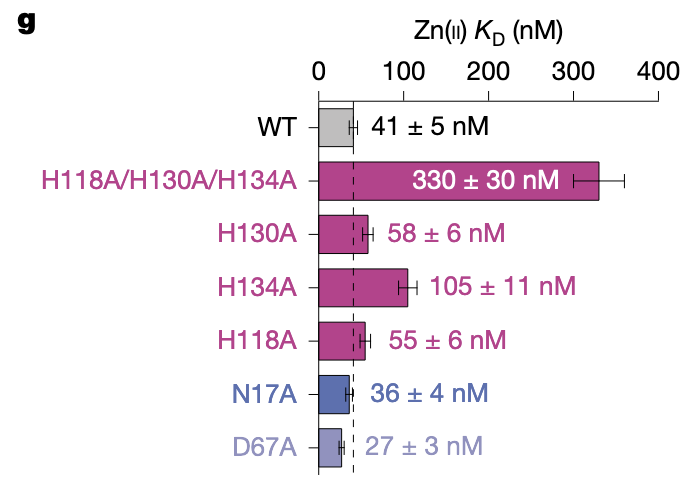
野生型 KD ≈ 41 nM——不是天然金属酶那种超强结合(<10 nM),但已经非常不错。
这里的数字透露了一条信息:
ZETA_1 是真正意义上的“新生金属口袋”,还不具备天然酶那种极端优化过的金属亲和力。
这反而增加了它的魅力——它还可以更强。
(5)Fig.3h–i:逐个拔掉催化残基,看它会发生什么
突变实验是检验设计逻辑是否自洽的终极方式。
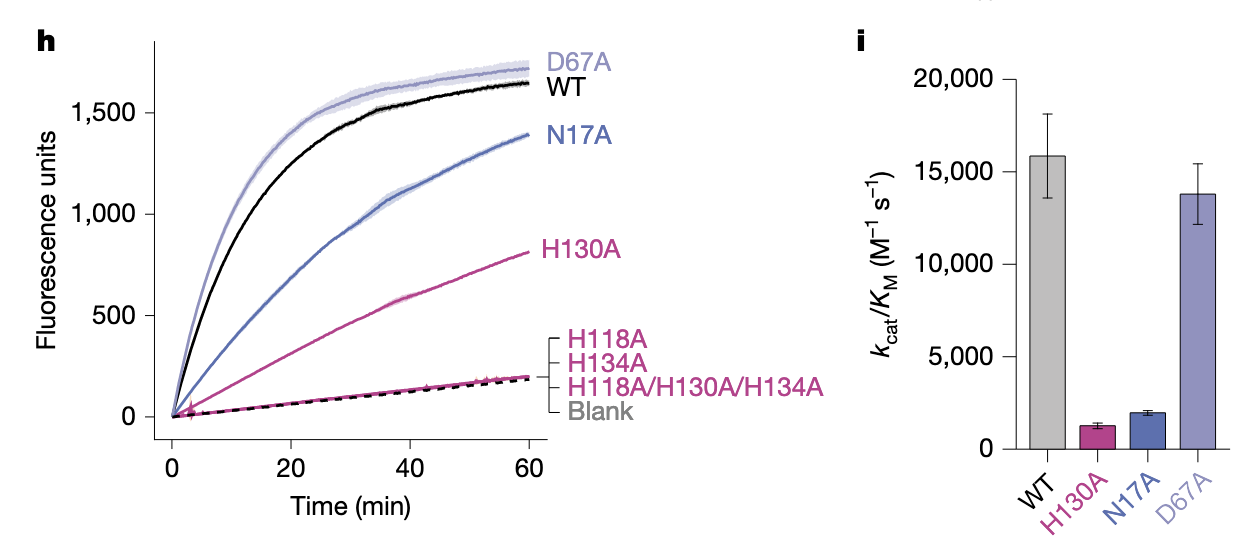
● 三个 His → Ala:
完全失活。说明模型对配位几何的安排是对的。
● D67A:
没有降低活性,甚至让 Zn²⁺ 结合更好。
这是一个非常有趣的结果——
暗示 D67 可能并不是主要 general base,
甚至可能与 H130 争抢金属。
这条线索在后面的 ZETA_2、ZETA_3 设计中发挥了巨大作用。
● N17A:
活性降低约八倍,说明它的氢键确实稳定了底物。
这一系列突变实验给出了一个非常有力的结论:
ZETA_1 的活性不是“偶然”,而是由设计出的几何严格决定的。
它是一条“有理由”的酶。
Figure 3给出了一个完整的叙事闭环:
模型设计的 pocket 是真实可折叠的;
真实 pocket 的原子位置几乎与设计模型一致;
活性来自金属和几何,而不是“偶然变构”;
突变实验揭示了真实的反应机制;
ZETA_1 不是自然界的仿制品,而是一条真正“人工创造的催化实体”。
你会发现这篇论文用一种非常克制的方式传达着一件震撼的事实:
我们第一次看到了一条从理论几何 → AI 生成 → 实验折叠 → 精准催化全链路都自洽的 de novo 金属水解酶。
而 Fig.3,是这条链路的“现实验证”。
六、第二轮设计:更准确的催化假设,更高的成功率(Fig.4)
第一轮设计中,ZETA_1 给了一个非常重要的反馈:
D67 的角色可能并不像最初假设的那样明确,它甚至可能与 H130 争夺 Zn²⁺。这个现象提示了一个关键点——如果希望提升催化效率,应该更明确地在设计中加入“真正的 general base”。
第二轮设计就从这里出发。
(1)更清晰的 theozymes:把 general base 显式纳入 DFT 模型(Fig.4a)
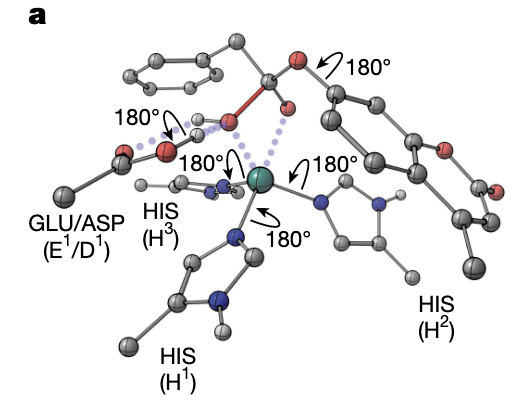
Fig.4a 显示新的 DFT 过渡态模型,这一轮最重要的升级是:
催化碱(Glu/Asp)不再是“推测出来的”,而是被直接固定在理论几何中;
Zn²⁺ 配位几何更明确;
三个 His 的 Cβ 位置也被建模,使得生成结构时对侧链方向的要求更合理。
这一改变让输入 motif 更接近天然金属酶的布置方式,也让 RFdiffusion2 能够围绕一个“更精确的催化核心”生成结构。
(2)结果的变化非常明显:成功率显著提升(Fig.4b)
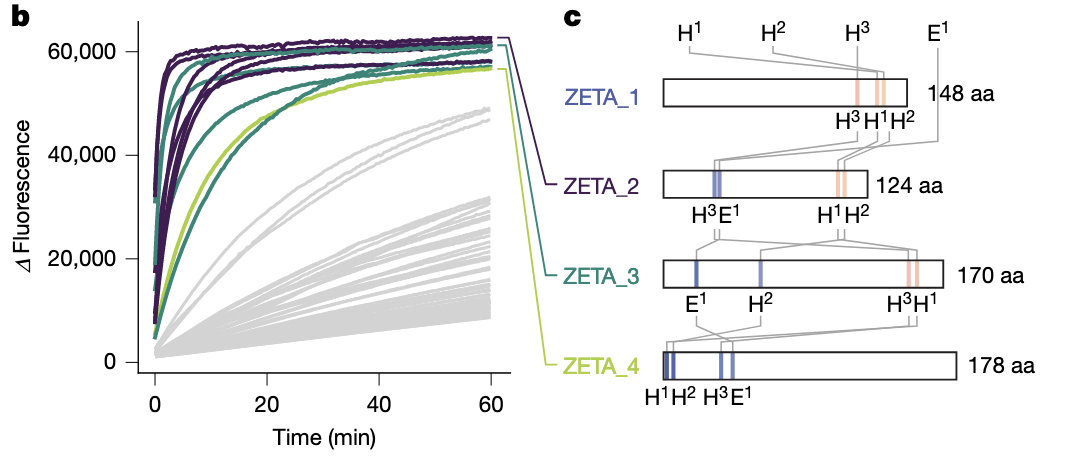
Fig.4b 展示的是第二轮 96 个设计的反应曲线。
第一轮你会看到只有五条曲线上升得明显,而本轮:
有 11 条设计表现出清晰的酶活性;
这些活性来自 3 种完全不同的折叠类型(scaffold families)。
这说明 RFdiffusion2 在第二次输入更清晰的催化需求后,
能在结构空间中找到更多“可用、有效、稳定”的路径。
简单说:
输入 motif 越准确,模型越容易找到可行解。
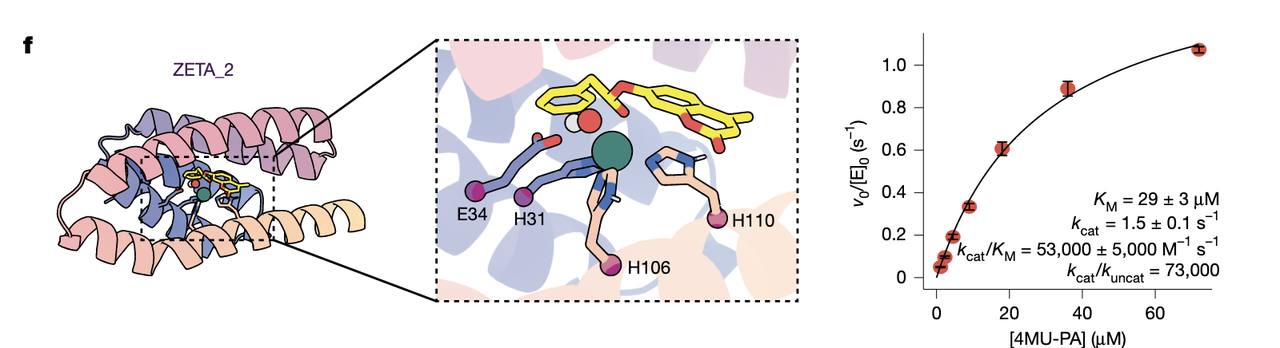
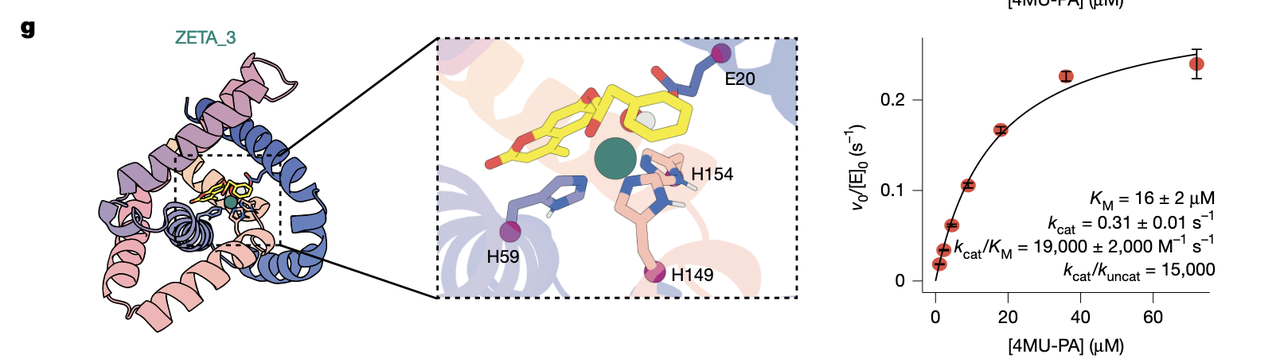
(3)ZETA_2、ZETA_3、ZETA_4:三种不同折叠,三种催化方案(Fig.4c)
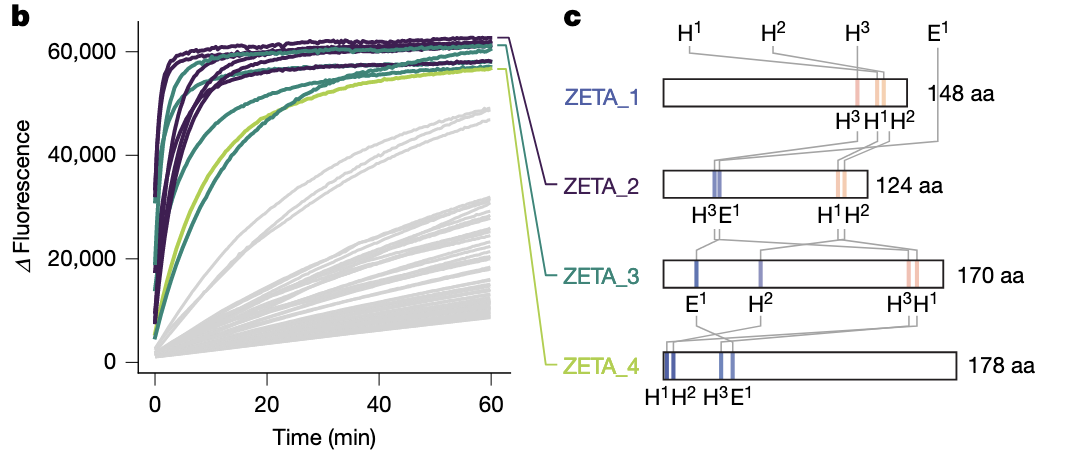
Fig.4c 这张图非常值得注意。
它展示三个最高活性设计的序列长度与催化残基在骨架中的位置。
你会发现:
ZETA_2:124 aa,三 His 和 general base 的排列非常紧凑
ZETA_3:170 aa,折叠较大,催化残基明显分散
ZETA_4:178 aa,结构更复杂,口袋形状也不同
三个酶的骨架完全不同,但它们都能稳定地再现相同的催化几何。
这再次支持前文的核心观点:
RFdiffusion2 不再依赖“模板折叠”,而是围绕催化几何自由创造结构。
(4)动力学数据(Fig.4d–e):性能全面超过第一轮
Fig.4d–e 给出了所有 hits 的总体表现。
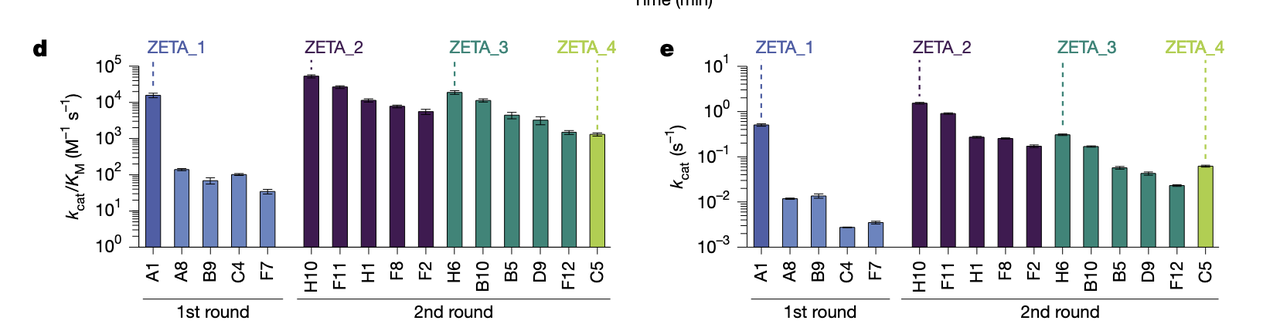
其中最引人注意的是:
● ZETA_2:kcat/KM = 53,000 ± 5,000 M⁻¹ s⁻¹
比 ZETA_1(16,000)再提升 3 倍
kcat 达到 1.5 s⁻¹,几乎是天然金属水解酶的水平
● ZETA_3:19,000 M⁻¹ s⁻¹
接近第一轮的 ZETA_1,但底物结合模式完全不同。
● ZETA_4:1,300 M⁻¹ s⁻¹
虽然活性比前两者低,但仍是传统 de novo 金属酶的高水平。
更重要的是第二轮的整体活性分布——
绝大多数 hits 活性都在 10³–10⁴ 量级,这说明:
设计流程已经可以稳定地产生高活性金属酶,而不是依赖偶然性。
(5)Fig.4f–h:三个代表设计的活性口袋长什么样?
这三张局部结构图展示了三种典型的催化策略:
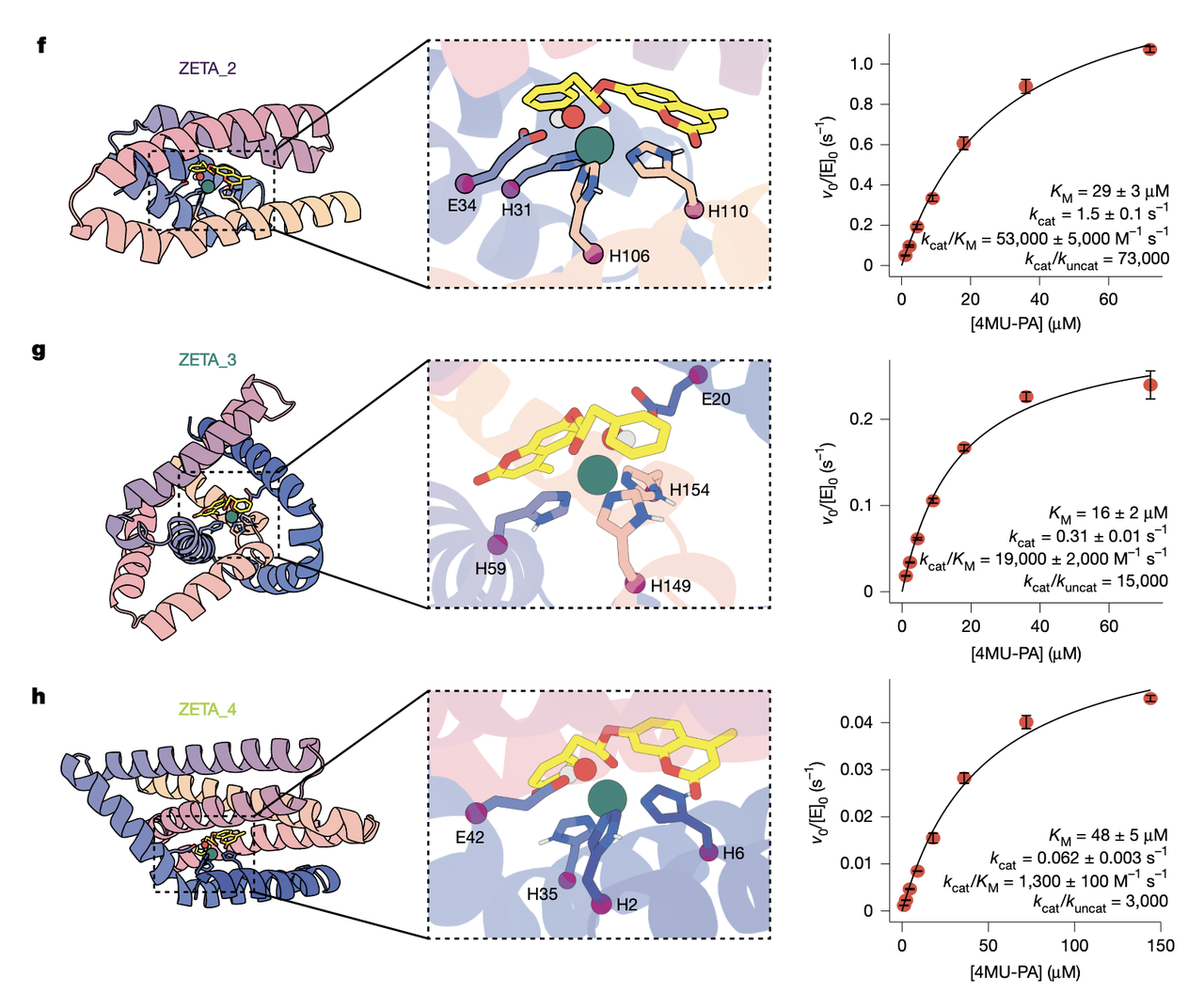
● ZETA_2
general base(Glu)位置极佳
Zn²⁺ 配位三 His 形成一个非常干净的三角几何
底物的 coumarin 环系展露在溶剂侧
ZETA_2 的结构给人的感觉是“精准且紧凑”。
● ZETA_3
与 ZETA_2 方向相反:PA 部分暴露在外
His 排布更松散,但 pocket 更深
可能通过更强的疏水约束定位底物
这也需要结合 Extended Data Fig.7(下一节会讲),才能更好理解它与 ZETA_2 的差异。
● ZETA_4
结构最复杂,口袋较大
几何上略显宽松,但仍能稳定过渡态
ZETA_4 像是“更通用的口袋”,虽然效率不如前两者,但结构空间展示了更大的灵活度。
(6)第二轮的核心意义
这一部分不需要太多渲染,结论本身已经很清晰:
更准确的催化 motif → 模型能找到更多可行结构
RFdiffusion2 的结构生成不是孤立事件,而是可迭代、可收敛的
设计出的酶活性已经进入天然金属酶的区间(10⁴–10⁵ M⁻¹ s⁻¹)
而结构仍然完全是 de novo 的,不模仿自然界
这说明:
我们正在见证一种“计算驱动的催化性能提升曲线”,类似自然界的进化,但速度是迭代模型的速度。
七、晶体结构验证:ZETA_2 真的按 AI 的想象折叠了吗?(Fig.5)
在酶设计文章里,动力学数据能告诉你“它动了”,突变实验能告诉你“它按设计那样动”,但只有晶体结构能回答最核心的那个问题:
AI 所构建的结构,在现实世界是否真的以相同的原子排列方式存在?
Fig.5 就是在回答这个问题。而它给出的答案清晰得几乎令人意外。
(1)Fig.5a:整体折叠几乎一模一样
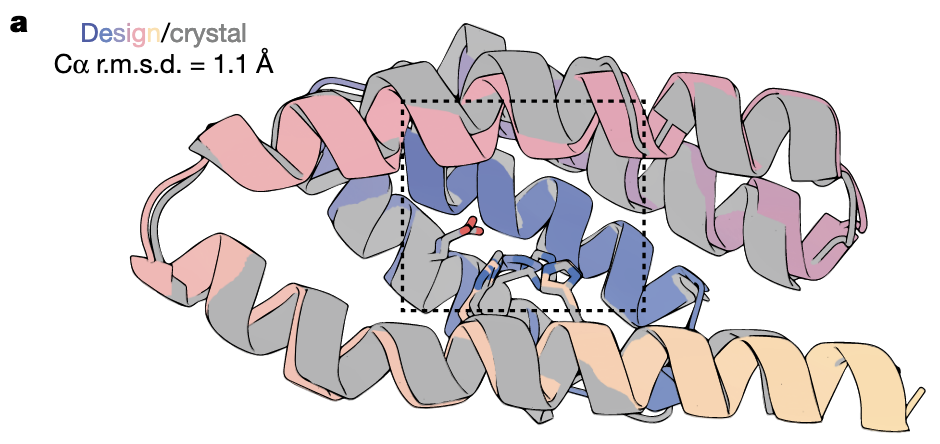
图上是设计模型(彩色)与晶体结构(灰色)的 Cα 对齐。RMSD = 1.1 Å。
这是一个极低的数字。
在结构生物学里,只要两个结构 RMSD < 2 Å,就可以认为“折叠一致”。
而 1.1 Å 代表的是:
不仅整体折叠一致,连局部构象都高度吻合。
换句话说,RFdiffusion2 并不是生成一个“能折叠大概成那样”的蛋白,而是生成一个“现实中确实会折成那个样子”的蛋白。
在 de novo 场景中,这是非常罕见的。
(2)Fig.5b:活性位点的原子排布也在正确的位置
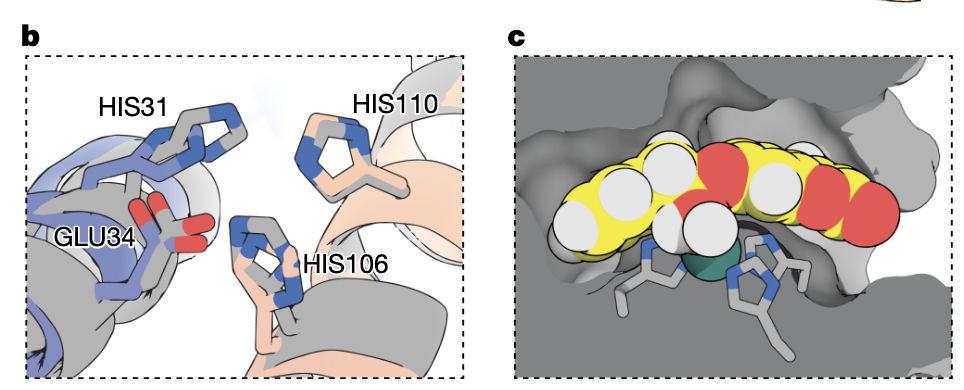
在 Fig.5b 的局部放大中:
三个 His 配位 Zn²⁺ 的方向一致;
General base(Glu)的位置恰在应在的位置;
周围 pocket 的形状几乎照搬了设计模型。
这说明 AI 的设计不仅是“折叠正确”,而是催化几何也正确。
要知道,催化位点通常是设计中最容易偏移、最容易塌陷的部分,而这里却精确到能与 DFT 过渡态完美对接。
这是一个强烈的信号:
RFdiffusion2 不只是设计结构,而是在设计“功能性结构”。
(3)Fig.5c:口袋形状与过渡态模型完全互补
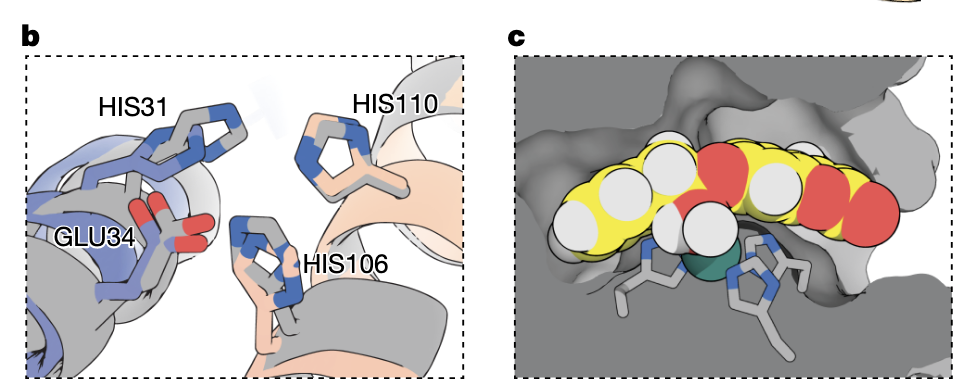
这一帧非常值得强调。
它展示了设计时使用的过渡态模型(transition state analogue)叠加在晶体结构 pocket 里。
你会看到:
底物的定位空间几乎与设计模型重合;
口袋壁的形状对称、紧密,并无塌陷或意外的张力;
活性位点留出的空间恰到好处,不多也不少。
这张图的美学意义不亚于科学意义:
它展示了一种“从功能出发的形状”,
AI 给了它最合理的结构支撑,
而自然界认可了这个形状。
这是第一次,一个完全不存在于自然界的金属水解酶,在现实世界以原子级别重现了它在计算空间的预测结构。
(4)晶体结构背后的两层重要结论
从 Fig.5 提炼出的两层深意:
第一层:AI 已能够生成“可折叠、可结晶”的 de novo 酶骨架
天然蛋白质之所以能结晶,是因为它们的结构稳定、重复性好、构象分布窄。
ZETA_2 能被结晶,说明:
折叠能量面简单;
活性位点稳定;
整体结构没有显著的柔性导致 X 射线衍射模糊。
它是一条“成熟的蛋白”,而不是一条“勉强折起来”的设计品。
第二层:金属配位几何的准确性验证了整个 RFdiffusion2 设计逻辑
金属酶的配位几何极为挑剔——稍偏几度几 Å 都可能让催化失效。
晶体结构的吻合度说明:
RFdiffusion2 内部确实在围绕催化几何展开结构生长,而不是硬塞残基。
这与第一轮的 PLACER 分析(Fig.2e–h)以及突变数据(Fig.3h–i)形成了一个完整的闭环。
(5)为什么 Fig.5 可以看作全篇的“验证之锤”?
因为只要这一张图成立,整篇文章的所有设计链路就成立:
DFT 的催化要求有意义
RFdiffusion2 能根据催化几何生成可折叠结构
ProteinMPNN 的序列赋形有效
AlphaFold2 预测可信
PLACER 评估活性位点预组织是有用的
实验得到真实的高活性酶
晶体结构确认了所有计算的逻辑链条
从一个研究者的视角,Fig.5 对这篇文章的价值几乎等于一句话:
这是一个真正从理论 → AI → 实验 → 结构验证 全链路跑通的金属酶设计案例。
它标志着金属酶设计从“可能做到”走向“确实可行”。
八、从 ZETA 系列到未来:金属酶设计的逻辑链终于闭合了
当我们读完 ZETA_1 的催化数据、ZETA_2 的晶体结构,以及第二轮设计的全面“提速”,就会发现一个事实已经悄悄发生:
金属酶设计,这个被认为最难的酶设计方向之一,第一次出现了“从计算机直接拿到高活性酶”的案例。
过去要做一条新金属酶,需要经历的步骤非常长:
先要手工定义活性位点 → 选折叠模板 → 插入残基 → 修 geometry → 调整 pocket → 表达失败 → 重来 → 勉强得到活性 → 定向进化若干轮……
这一切在 ZETA 系列中被彻底颠覆。
(1)RFdiffusion2 的本质突破:让“功能”成为蛋白质折叠的起点
文章结尾强调了一个核心思想:
RFdiffusion2 不再需要你告诉它“哪个残基是 His、要放到序列第几位”,
它只需要知道“催化必须的原子怎么放”。
蛋白质的骨架、折叠方式、序列——这些在传统设计中是“输入”,
在 RFdiffusion2 里全部变成了“自由变量”。
而唯一固定的,是那团被 DFT 算出来的催化几何。
这是一种从根本概念上重新思考蛋白质设计的方式:
不再是“我想造一条蛋白质,让它顺便催化反应”,而是“我要达成这个催化几何,蛋白质应该如何生长?”
当催化要求变成结构生成的“种子”,设计逻辑就反过来了。
而这种反转,是 ZETA 系列能一上来就高活性的真正原因。
(2)PLACER 和 Chai-1 的加入:让“选择正确”成为可能
过去我们经常会问:
设计出来的几十条候选,哪些值得投入实验资源?
文章在第一轮和第二轮 hits 的对比中给了一个惊人的线索——
PLACER 对“活性位点预组织化”的预测,几乎直接对应实验结果。
也就是说:
活性高的 pocket,在 PLACER 中几乎“不动”;
活性弱的 pocket,底物在模拟中“到处乱跑”。
这让我们第一次拥有一种近似“能看懂催化几何是否稳定”的 ranking 方法。
Chai-1 也补了一刀:
它可以在 pocket 内预测 Zn²⁺ 和底物的结合方式,从中判断 Zn-binding 是否有异常构象(例如 ZETA_1 中 H130 vs D67 的竞争)。
这些工具组合在一起,意味着:
我们不只是在设计结构,而是在设计能通过“分子力学筛查”的功能性结构。
这是金属酶设计稳定化的关键节点。
(3)第二轮设计的意义:证明“改 motif → 效果立刻改善”的迭代能力
在第一轮中,作者注意到 general base 的角色不够明确,于是他们修改了 DFT 输入,重新训练/推理,得到:
活性提升至 53,000 M⁻¹ s⁻¹(ZETA_2);
更多 hits;
不同的折叠都能实现同一个催化几何;
甚至能通过 ORI token 控制底物的取向。
这说明一个非常重要的事情:
金属酶设计进入了可控迭代期。
你只要让 DFT 或 QM 给出更精准的几何,RFdiffusion2 就能找到更好的结构响应。
这几乎把传统的“需要几个月才能进行一次的蛋白工程迭代”转换成“几小时就能完成一次 motif 调整和结构生成”。
金属酶的设计速度因此进入“量级提升”。
(4)晶体结构的终极意义:证明这是“工程”,不是“幸运”
ZETA_2 的晶体结构是全文的定点时刻。它在 1.1 Å 水平上重现了 AI 的预测——这意味着:
模型在处理金属配位几何时是可信的;
设计出的 backbone 真的是最低能量构型之一;
活性位点的约束足够强,使 pocket 能以近乎同一构象呈现。
这里的验证不是局部的,而是“整体结构 + 活性几何”双重一致。
因此这不是幸运事件,而是一个成体系的成功。
(5)文章最后的“预言”:金属酶的可编程时代正在来临
作者在结尾写得非常克制,但含义深远:
RFdiffusion2 + PLACER 的组合不仅能设计 Zn 酶;
它也适用于更多类型的金属催化和复杂反应;
甚至适用于非金属催化,例如 serine hydrolase(他们在引用里提到了自己的另一篇工作)。
真正的意思是:
我们正在进入“用几何定义反应,用 AI 生长结构”的时代。
酶的反应类型,将不再受限于自然选择给我们的模板。
这句话在金属酶领域尤其重要——
金属酶往往催化最难的反应,也是环境处理、材料降解、化学合成中最需要的酶类。
现在它们终于变得可被“设计”。
(6)总结整篇文章最核心的三条 take-home message
如果只带走三件事,我会这样写:
① 催化几何是核心,蛋白质只是满足几何的结构载体。
RFdiffusion2 把酶设计从“造蛋白”变成“造催化场景”。
② 预组织化决定活性。
PLACER 的分析重复强调:稳定的 pocket 才是高活性的根本。
③ 金属酶设计的迭代速度,将从多年缩短到数天。
DFT motif 越好,设计结果越好;
模型不依赖模板折叠;
成功率随着 motif 优化呈线性提升。
这是一个新的酶设计范式。
结语:当催化几何变成种子,蛋白质设计的边界被重新划定
当催化几何变成种子,蛋白质设计的边界被重新划定
读完整篇文章,最强烈的感受不是“AI 又做到了什么”,而是金属酶设计这个长期被认为几乎无法系统解决的问题,正在第一次呈现出一种可被工程化、可被迭代、可被预测的形态。
这一点在 Fig.5 的晶体结构中达到巅峰——当 ZETA_2 的原子在现实里以 1.1 Å 的精度对齐计算模型,你会意识到:
AI 不是在“猜一个结构”,它是在“构建一个满足催化要求的解”。
这意味着一个新的逻辑链已经真正闭合:
催化几何由量化计算给出;
RFdiffusion2 围着几何生长出可折叠的骨架;
MPNN 赋予序列;
AlphaFold 与 PLACER 评估可行性;
实验确认折叠与活性;
晶体结构给予最终验证。
过去十几年,酶设计的主旋律是“找一个折叠 → 塞进催化残基”。
而这篇 Nature 展示的是完全相反的方向:
先点亮反应的核心原子,结构则围着功能长成。
这不是一次方法学的升级,
而是一次设计哲学的迁移。
从“造蛋白”到“造催化场景”,
从“模板导向”到“几何导向”,
从“希望它折出来”到“它本来就会折出来”。
如果说 ZETA 系列证明了什么,那就是——
金属酶设计不再是“非自然界不可”,
它开始成为一件人类能够系统、快速、主动完成的工程。
而这,可能正是未来十年蛋白设计领域最值得期待的转折点。
延伸阅读
本文属于 AI4S文献 栏目。
返回 AI4S文献 → 去公众号阅读完整版 →