够简单,也够强大:苹果发布蛋白结构预测工具 SimpleFold
今天突然发现苹果也开始做蛋白质预测了,相当震惊,于是读一下苹果的蛋白质预测,甚至是生物学方面的处女作。
一、背景
在蛋白质结构预测领域,大家最熟悉的名字无疑是 AlphaFold2 和 RoseTTAFold。这两类模型在方法论上有一个共同点:依赖大量的领域特定设计,例如 多序列比对(MSA)、pair 表示、以及 triangle updates 等复杂模块。这些设计的初衷,是把我们对蛋白质结构形成的先验知识“硬编码”进模型里,从而提升准确性。
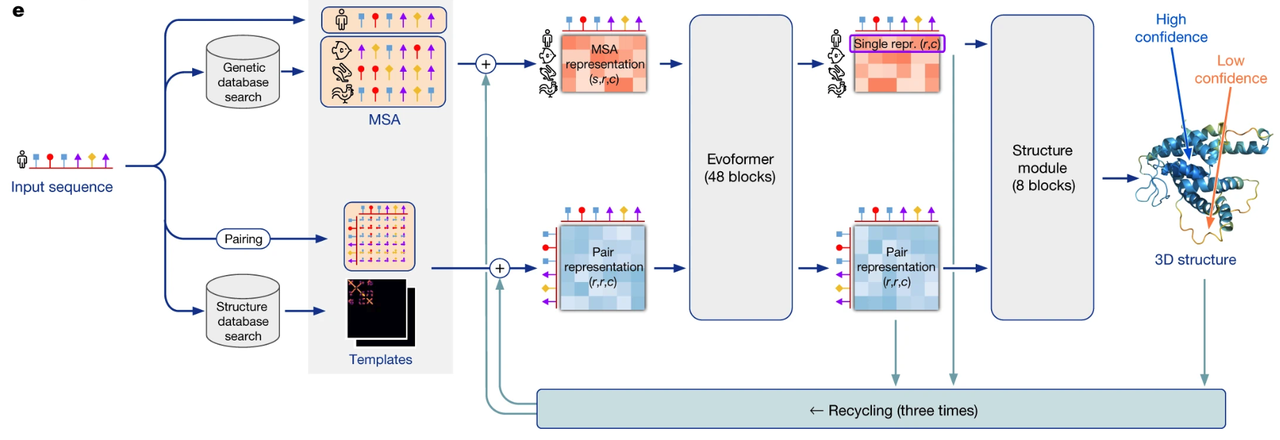
今天惊讶的发现:苹果公司也开始做蛋白质折叠预测了。没错,就是卖手机卖电脑的巨头苹果。他们发布了一个名为 SimpleFold 的模型。顾名思义,这个模型最大的特点就是“简单”——它几乎抛弃了前人构建的各种复杂结构,仅仅用标准的 Transformer 堆叠起来,却依然能达到接近甚至超越主流模型的表现。

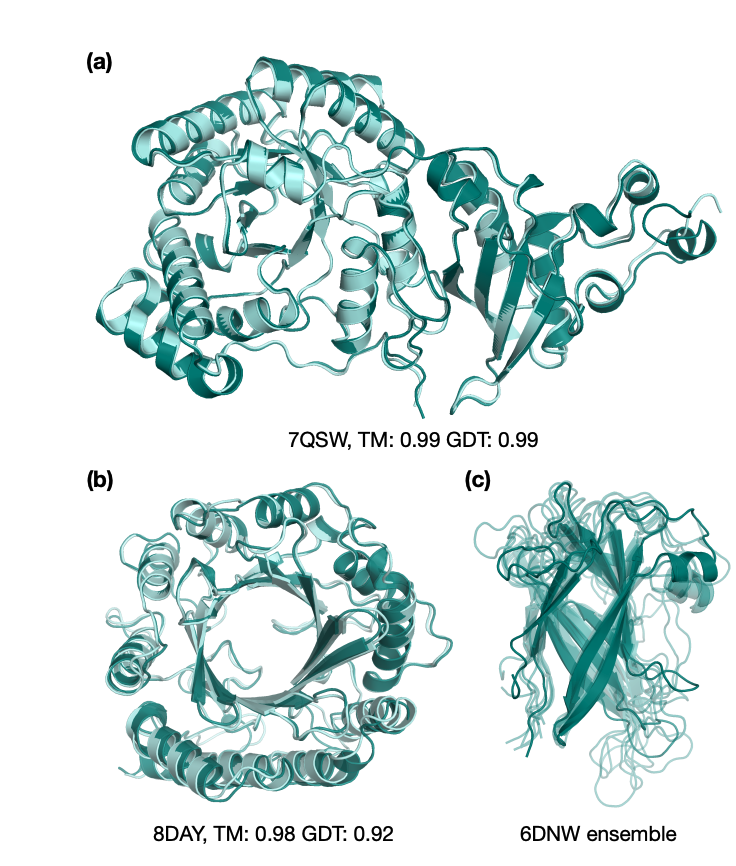
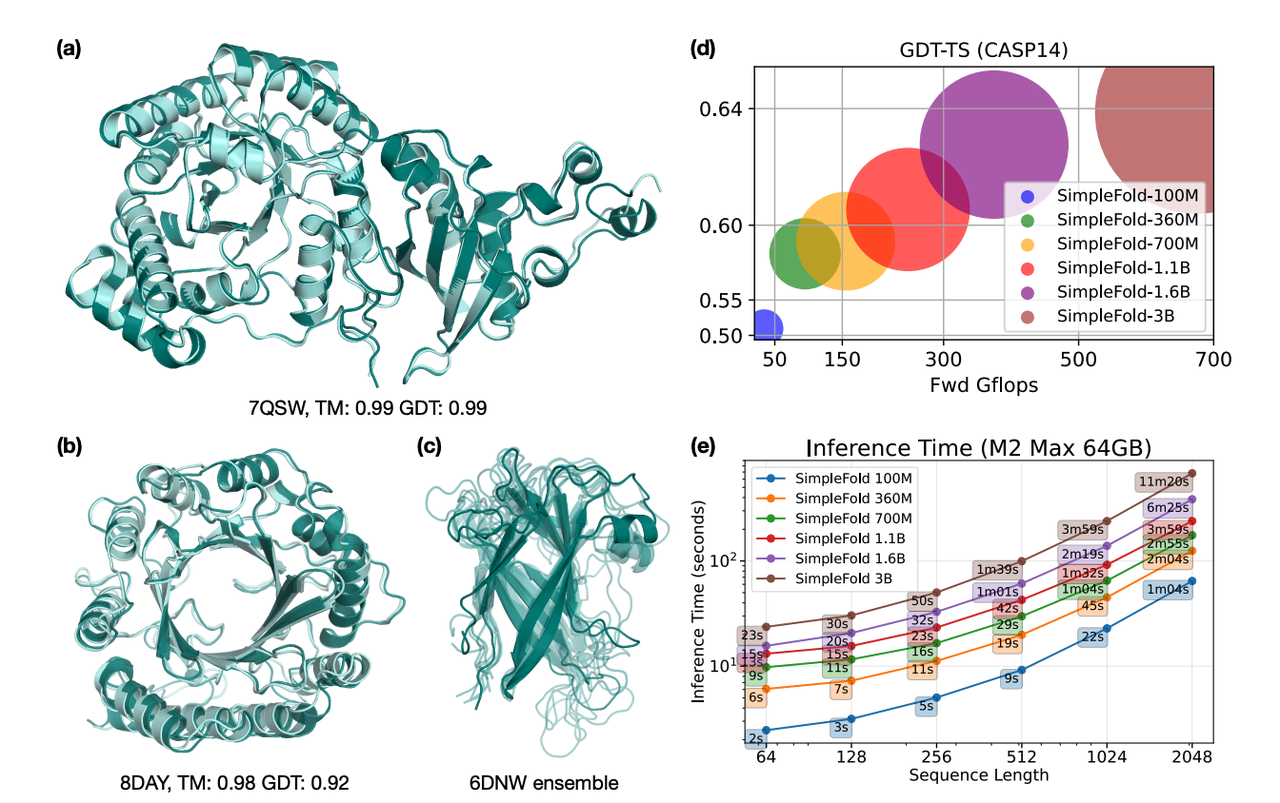
如果你先看文章中的图 1(a)(b),就会更直观地感受到这个结果:图中展示了 SimpleFold 在两个目标蛋白(7QSW 和 8DAY)上的预测结构(深蓝色)和实验结构(浅蓝色)的对比。两者几乎完美重合,说明即便没有繁琐的模块,SimpleFold 依旧能给出高度准确的预测。
二、研究出发点
在过去几年里,蛋白质折叠预测的突破主要来自 高度定制化的架构设计。比如 AlphaFold2 引入了多序列比对(MSA)来捕捉进化信息,用 pair representation建模残基之间的相互作用,再通过 triangle updates 模块来强化几何约束。
这些设计虽然有效,但也带来两个问题:
计算昂贵 —— 运行一次完整预测往往需要大量算力;
泛化受限 —— 模型对孤儿蛋白(几乎没有同源序列的蛋白)往往表现不佳。
苹果团队的思考很直接:
这些复杂的领域特定模块,真的必不可少吗?
他们的答案是 —— 不一定。与其依赖“人类专家的手工设计”,不如借鉴在图像、文本生成领域已经被验证有效的思路:完全用通用的 Transformer 架构,通过大规模数据和生成式训练目标,让模型自己学习蛋白质的对称性和规律。
这就是 SimpleFold 的设计理念:
不用 MSA
不建 pair representation
不做 triangle updates
完全依靠 Transformer + flow-matching 生成目标
如果你对照文章中的 Figure 2,会更清晰地理解这种“简化”思路。图中展示了 SimpleFold 的核心架构:
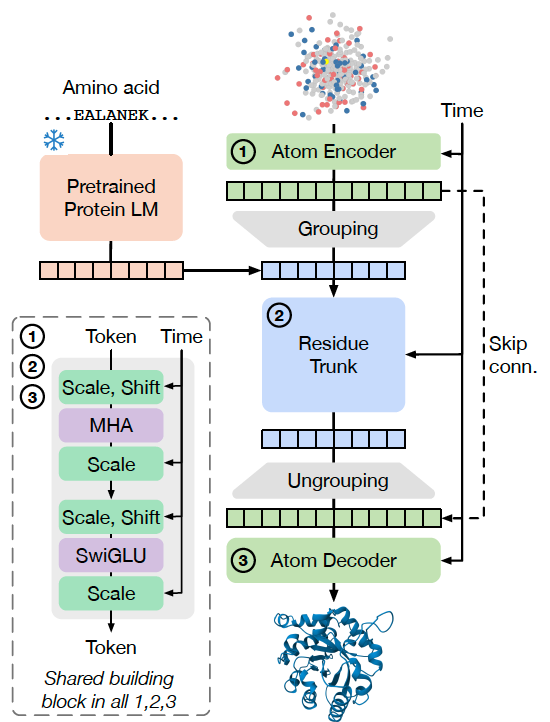
左侧是 原子编码器,负责将输入的“噪声原子坐标”转化为特征;
中间是 残基主干(Residue Trunk),承担主要计算任务;
右侧是 原子解码器,将特征还原为 3D 结构。
整个流程只用了一种通用的 Transformer block,没有任何 AlphaFold2 那种复杂的几何模块。
这意味着:SimpleFold 重新定义了蛋白质折叠问题,把它当成一个“条件生成任务”——从氨基酸序列出发,生成原子级别的三维结构。
三、方法核心
- 生成式建模思路
传统模型大多把折叠当作“回归问题”——给定氨基酸序列,直接预测对应的结构。
SimpleFold 则换了一个角度:把折叠当作生成任务。
输入:氨基酸序列,相当于生成模型的“条件”
输出:完整的原子级三维坐标,相当于“生成的结果”
过程:从高斯噪声出发,逐步“流动”到合理的蛋白质结构,这就是 flow-matching 的核心思想。
这个思路和图像生成(text-to-image)、三维生成(text-to-3D)如出一辙,只不过这里生成的对象是蛋白质结构。
- 模型架构
在 Figure 2 里,作者展示了 SimpleFold 的整体框架:
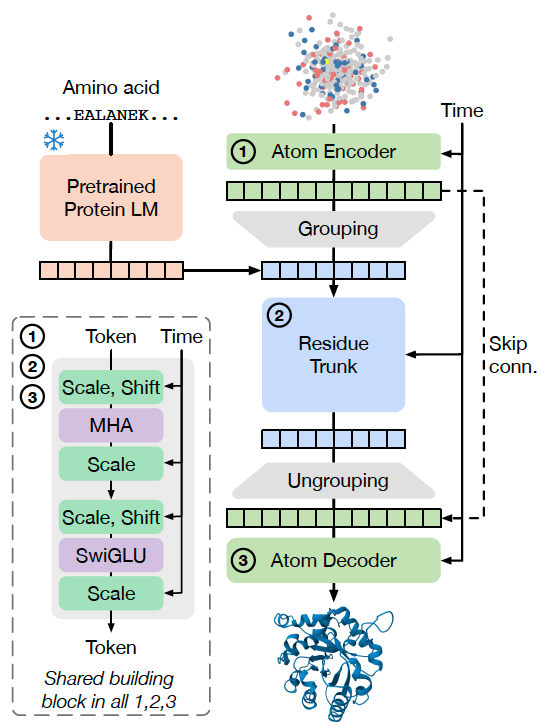
- 原子编码器 (Atom Encoder):
将带噪声的原子坐标和基本原子信息(类型、电荷等)转化为特征表示。
- 残基主干 (Residue Trunk):
模型的核心部分,基于标准 Transformer 进行序列建模;
同时引入了 蛋白质语言模型 (ESM2-3B) 的 embedding 作为条件输入。
- 原子解码器 (Atom Decoder):
将残基级别的特征还原到原子层面,输出预测的结构坐标。
与 AlphaFold 系列最大的不同在于:
没有 pair 表示
没有 triangle updates
也没有专门的等变(equivariant)几何模块
模型仅依赖 Transformer 的自注意力机制和数据驱动的学习。
- 数据与规模
SimpleFold 之所以能站得住脚,核心在于 大规模训练数据:
PDB 实验结构:约 16 万条
AFDB 和 ESM Atlas 的蒸馏数据:数百万条
最终,最大模型 SimpleFold-3B 用了 约 900 万个结构进行训练,参数量达到 30 亿。
这种规模的训练,使得即使架构简化,模型依旧能学习到复杂的折叠规律。
对应 Figure 4 的 scaling 曲线可以看到:随着参数量和数据量增加,SimpleFold 的表现稳步提升,尤其在复杂任务上,3B 模型的提升非常明显。
小结
SimpleFold 的方法核心可以概括为三句话:
用 flow-matching,把折叠当作生成任务;
架构极简,仅由 Transformer 组成;
依靠大规模训练数据与模型规模扩展,保证预测精度。
四、性能与结果
- 基准测试表现
在两个常用的折叠基准上,SimpleFold 都展现出强劲的表现:
CAMEO22:这是一个持续更新的蛋白质结构预测评测平台。SimpleFold 在这里取得了与 ESMFold、AlphaFold2 等主流模型接近的成绩,部分指标甚至超过了 ESMFlow(同样是生成式方法)。
CASP14:更具挑战性的盲测基准。SimpleFold-3B 的成绩明显优于 ESMFold,并在多个指标上接近 AlphaFold2 和 RoseTTAFold2。

如果你看文章中的 Figure 1(d),会发现模型规模和性能几乎呈现线性关系:从 100M 到 3B 参数,模型越大,性能越强。这说明 SimpleFold 的“简化架构”并没有成为瓶颈。
- 真实案例对比
论文中的 Figure 1(a)(b) 给出了两个具体蛋白的预测:
7QSW(RubisCO 大亚基)
8DAY(Dimethylallyltryptophan synthase 1)
图中浅蓝色是实验结构,深蓝色是 SimpleFold 的预测,几乎完全重合(TM-score > 0.98)。
这直接说明:即使没有复杂模块,SimpleFold 的预测依然可以非常精准。
- 多构象与柔性建模
传统回归式模型往往只能输出“单一最优构象”。而 SimpleFold 由于采用生成式目标,天然能生成 多样的合理构象,这在药物发现等场景尤其重要。
Figure 1(c) 展示了一个 Flagellar hook 蛋白(6DNW)的不同构象集合,SimpleFold 能够捕捉到其柔性变化。
在 ATLAS 分子动力学数据集和 Apo/Holo 任务上,SimpleFold 的多态生成效果超过了 ESMFlow、AlphaFlow 等方法,证明它在 建模蛋白质柔性和动力学方面有独特优势。
- 推理效率
另一个值得关注的点是 推理效率。
在 Figure 1(e) 里,作者展示了不同规模 SimpleFold 的推理速度;
即便是 3B 模型,在 MacBook Pro (M2 Max, 64GB) 这样的消费级硬件上也能运行;
更小的 SimpleFold-100M 模型在速度和准确性上达到很好的平衡(恢复了约 90% 的性能)。
这让 SimpleFold 更接近“普惠”科研工具,而不只是依赖超级计算机的实验室项目。
小结
从结果来看,SimpleFold 用“更简单的架构”达到了 接近最复杂模型的准确率,同时在 多构象预测和推理效率 上有额外优势。
五、意义与影响
- 方法论上的颠覆
过去几年,AlphaFold 系列几乎“定调”了蛋白质折叠预测的技术路线:依赖复杂的领域特定模块,比如 MSA、pair representation和triangle update。
而 SimpleFold 证明了另一条道路也同样可行:
抛弃复杂结构,完全依靠通用 Transformer;
依靠规模化训练,让模型直接从数据中学习蛋白质的几何规律。
这为未来的研究打开了新的思路:也许不需要生物学家“手工设计”的模块,通用大模型就能捕捉自然界的规律。
- 工程与应用价值
SimpleFold 的“简化”不仅是学术上的好奇心,更带来了实际好处:
计算效率更高:不再依赖昂贵的 MSA 搜索和复杂几何模块,推理速度更快;
部署门槛降低:在消费级硬件上也能运行,让蛋白质预测更普及;
多构象能力强:生成式框架天然支持蛋白质柔性和多态预测,对药物设计、结合口袋发现等任务意义重大。
- 行业层面的信号
苹果的入局本身也是一个强烈信号:
过去,蛋白质折叠预测的核心突破主要来自学术界和专门的研究机构(DeepMind、Baker 实验室等);
如今,苹果这样的科技巨头也投入其中,说明 蛋白质预测已不再是“科研小圈子”的事情,而正在成为通用 AI 的一部分。
这可能意味着未来:
工业界会更快把蛋白质预测模型应用到药物开发、材料设计等实际场景;
通用大模型的“跨界能力”将成为生命科学研究的一股重要推动力。
小结
SimpleFold 的重要性,不仅在于它的表现接近 AlphaFold2,而在于它 证明了复杂设计并非唯一解法,并且把蛋白质预测带向了“通用 AI 架构 + 大规模数据”的新范式。
六、总结
如果要用一句话概括 SimpleFold:
它用最简单的 Transformer 堆叠,实现了接近最复杂折叠模型的效果,还额外带来了多构象生成和更高效的推理。
这背后有三个值得记住的亮点:
架构简化:不再依赖 MSA、pair representation、triangular update,完全基于通用 Transformer;
生成式建模:通过 flow-matching,把折叠当作一个“从噪声到结构”的生成任务,自然支持多构象预测;
可扩展性与可用性:在大规模数据和参数扩展下持续提升,同时在消费级硬件上也能运行。
因此,SimpleFold 不仅是苹果在蛋白质设计领域的首次亮相,更可能代表一种新的范式:从复杂设计走向简洁通用,用数据和算力推动科学突破。
延伸阅读
本文属于 AI4S文献 栏目。
返回 AI4S文献 → 去公众号阅读完整版 →