原来水分子才是关键!新方法 WIIGA 显著提升蛋白–糖分子预测准确率
蓝极说:
在蛋白质设计和结构糖生物学领域,如何准确预测蛋白–糖分子的结合模式一直是一个难题:糖分子柔性高、结合位点浅表亲水,常常让传统对接方法“失灵”。

今天要和大家分享的是一篇来自阿根廷和意大利团队的最新工作——《Guided docking using solvent structure information improves the prediction of protein–glycan complexes》。作者提出了一种全新的引导对接策略(WIIGA),巧妙地利用蛋白质晶体结构中的水分子分布作为“线索”,显著提升了糖分子结合模式的预测准确率。
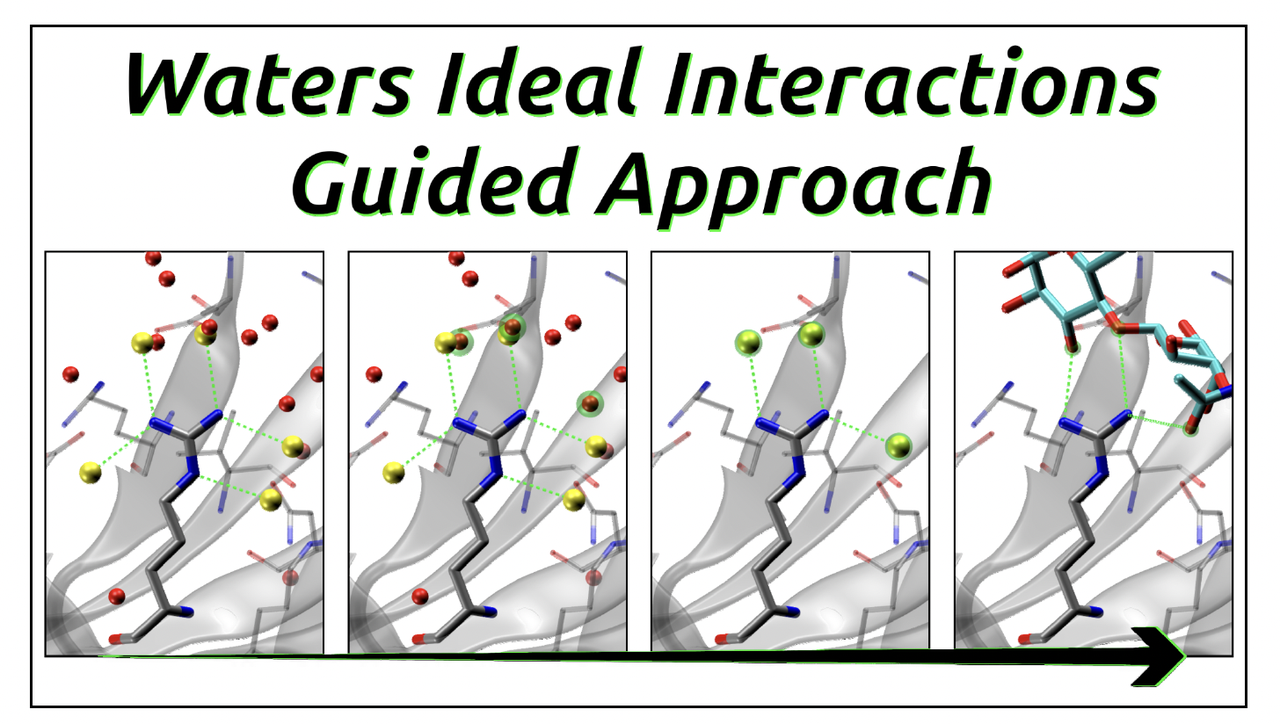
这项研究的贡献在于:
方法创新:把结晶水分子的信息转化为对接偏好,引导算法更接近真实结合方式;
效果提升:在大数据集上验证,比传统方法更准确、更可靠;
应用拓展:不仅能预测天然糖分子,还能应用到药物型糖模拟物,为未来的糖基药物设计提供了工具。
一:背景介绍
在结构糖生物学中,预测蛋白质–糖分子复合物的结合方式一直是难题。原因有两点:
糖分子柔性极高,可以摆出很多构象;
蛋白质与糖分子的结合位点通常是浅表而亲水的,不像小分子药物那样容易“卡”进去。
正因如此,虽然 PDB 中已经有 23 万多条蛋白质晶体结构,但真正包含蛋白-糖复合物的不到 2% 。
这篇文章的作者提出了一个新思路:
利用溶剂信息来引导对接。
为什么是水分子?因为在无配体的蛋白质晶体结构(apo 结构)中,结合位点往往会出现一些稳定的结晶水分子,它们的位置正好提示了未来糖分子可能与蛋白形成氢键或 CH–π 相互作用的“热点”。
📊 对应 Figure 1
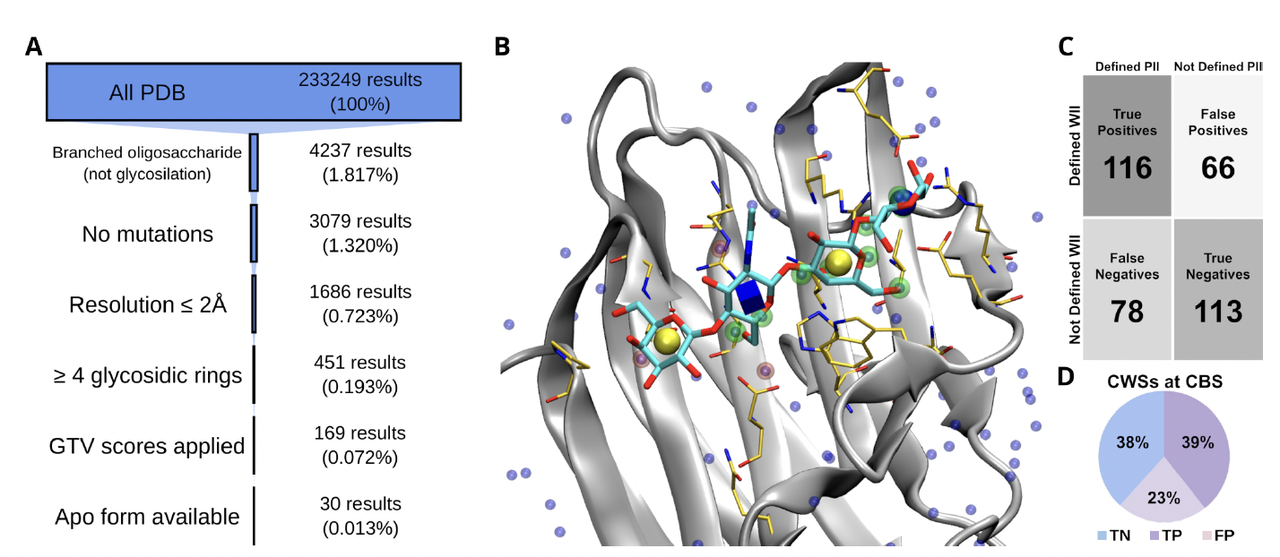
图 1A 展示了研究团队如何从 PDB 中筛选数据集,最终得到 30 个高质量的蛋白–寡糖复合物。
图 1B 则直观展示了一个例子:人类 Galectin-3 蛋白的结合位点上,apo 结构的结晶水分子(蓝色球体)与 holo 结构中糖分子的羟基位置高度吻合。
图 1C 和 1D 则进一步量化:并不是所有水分子都能成为有效的预测点,但有相当比例的水分子确实准确地“预示”了真实的蛋白–糖相互作用。
💡 要点总结:
研究的关键启发是——水分子的位置并不是随机的,而是蛋白质结合位点“记忆”糖分子结合方式的线索。这就是后续引导对接方法的核心思路。
二、方法核心:WIIGA 策略
解决蛋白-糖分子对接困难的关键,在于如何把 “水分子留下的线索”转化为算法可以利用的信息。作者提出了 WIIGA(Waters Ideal Interactions Guided Approach) 策略,具体分为四步。
📊 对应 Figure 2
图 2 直观地展示了 WIIGA 的流程,可以总结为:
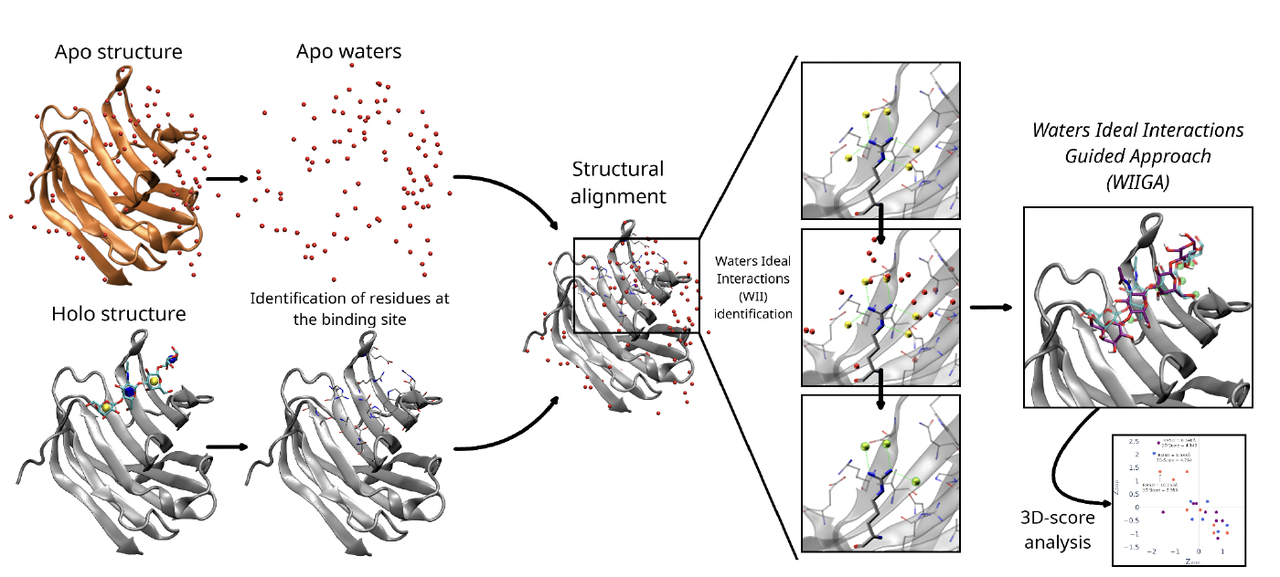
- 结合位点映射
先从有糖的 holo 结构里,找到与配体距离小于 5 Å 的残基,定义为 Carbohydrate Binding Site (CBS)。
然后把它和 apo 结构对齐,在 apo 结构中寻找与这些残基相关的结晶水分子。
理想相互作用 (Ideal Interactions, II) 的识别
对 CBS 残基,标出它们潜在的氢键受体/供体位置,或 CH–π 相互作用位置,这些位置就是“理想相互作用点”。
再筛选出与结晶水分子(CWS)位置接近的那一部分,称为 Waters Ideal Interactions (WII)。
- 引导对接
在对接过程中,把这些 WII 点设置成“奖励点”:如果糖分子的原子靠近这些位置,就会得到额外能量加分(–1.0 kcal/mol),提高该构象的排名。
可以把它理解成在结合位点里放了几个“路标”,引导糖分子朝着正确的方向结合。
- 结果评估
- 作者不只依赖能量打分,而是用 聚类 + 3D-score 的方法,把能量(Zene)和构象群体大小(ZPop)结合起来,挑选最合理的结合模式。
在 Figure 2 的图解里:
黄色球体代表 CBS 残基的理想相互作用点。
红色球体是 apo 结构中的结晶水。
紫色配体则是 WIIGA 预测出来的对接结果,可以看到它很好地贴合了参考 holo 结构的真实结合方式。
💡 要点总结:
WIIGA 的创新之处在于:不是盲目搜索,而是用“水分子提供的历史记忆”作为导航标志,引导糖分子找到正确的结合模式。
三、性能验证:WIIGA vs. 现有对接方法
蛋白-糖分子对接为什么难?原因在于:
传统方法(AutoDock Vina, Vina Carb, GlycoTorch Vina)主要针对小分子药物优化过,它们的打分函数不擅长处理糖分子柔性和亲水性结合。
结果就是:往往能生成一堆 plausible 姿势,但很难把真正的正确构象排在前面。
作者将 WIIGA 与 ADV(AutoDock Vina)、VC(Vina Carb)、GTV(GlycoTorch Vina) 进行了正面对比。
📊 对应 Figure 3
- Panel A:横轴是 RMSD(与真实结合方式的偏差),纵轴是正确预测的比例。
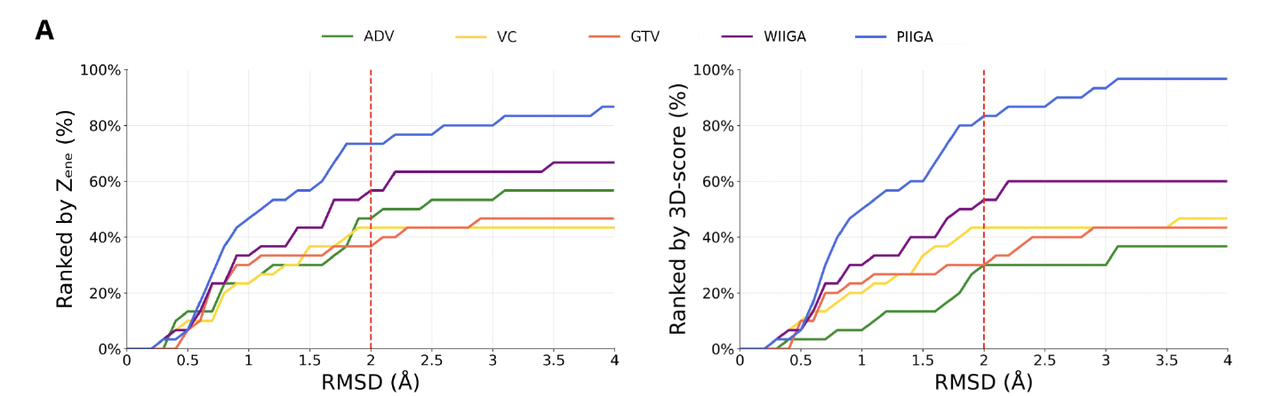
如果只用能量打分(左图),ADV 看似稍优。
但引入 3D-score 后(右图),WIIGA 的成功率明显领先:
WIIGA:53.3%
VC:43.3%
ADV/GTV:只有 30% 左右。
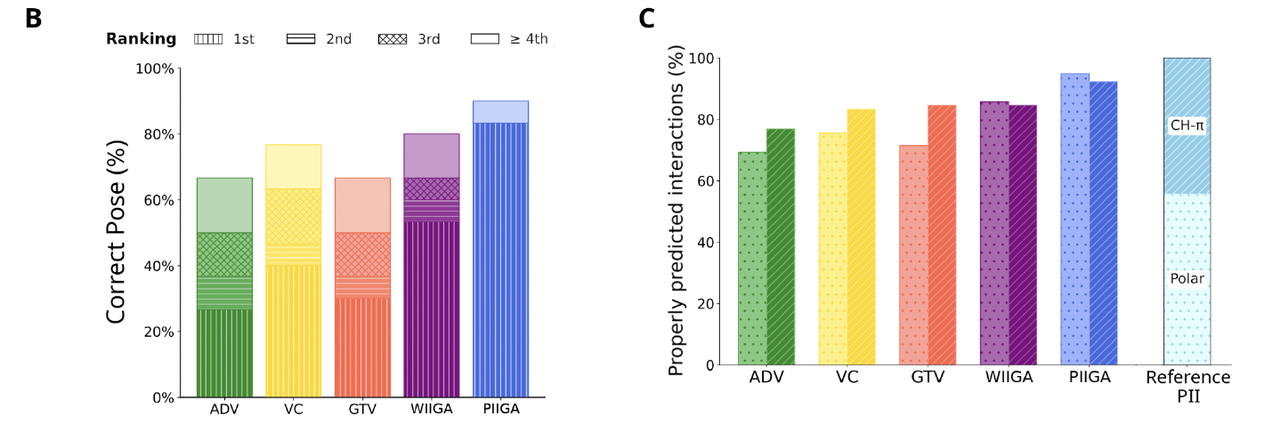
Panel B:统计了正确姿势(RMSD ≤ 2 Å)在结果排名中的位置。
- WIIGA 更常能把正确构象排在第 1 名,而不是被埋在一堆错误姿势里。
Panel C:比较预测结果中能否重现关键药效团相互作用(Pharmacophoric Ideal Interactions, PII)。
- 结果显示,WIIGA 更稳定地抓住氢键和 CH–π 相互作用,这恰恰是糖分子识别的关键。
💡 要点总结:
和传统方法相比,WIIGA 更擅长“抓住关键相互作用”。这使它不仅能生成正确的结合模式,还能把它们排在最前面,方便研究者一眼识别。换句话说,它提升的不是“可能性”,而是“可信度”。
四、3D-score:让选择正确构象更可靠
在对接预测中,常见的问题是:
能量打分往往不靠谱,尤其对糖分子这种柔性配体,能量差别可能很小;
结果就是一堆姿势都差不多,很难挑出真正正确的。
作者提出的 3D-score 就是为了解决这个问题。它结合了两类信息:
能量(ZEne):姿势本身的能量好坏。
群体(ZPop):这个姿势在模拟中出现的频率。
最终用公式 3D-score = –ZEne + ZPop 来综合判断。简单理解:
既要能量低,又要“大家都同意”它靠谱。
📊 对应 Figure 4
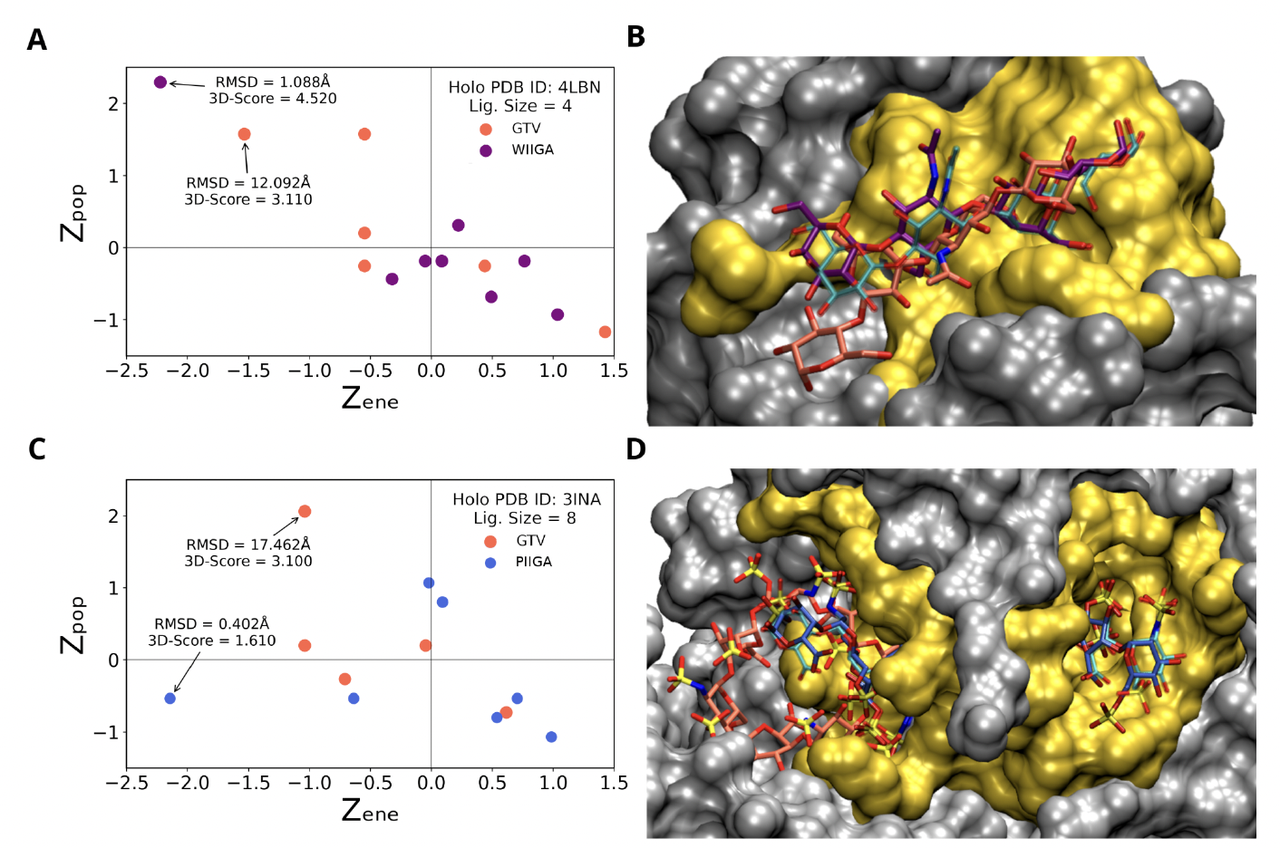
Panel A:散点图把所有姿势的 ZPop 和 ZEner 打出来,点越靠右上越好。可以看到 WIIGA 的预测结果明显聚在优势区域,而 GTV 的结果分散、难区分。
Panel B:对比可视化结构(人类 Galectin-3 CRD 结合乳寡糖),WIIGA 的预测(紫色碳原子)几乎与真实结合模式重合,而 GTV 的预测(橙色)明显偏差大。
Panel C & D:展示在更复杂的八糖体系中,3D-score 如何依然帮助从海量候选中识别出接近真实的构象。即便能量打分模糊,3D-score 依然能拉开正确解与错误解的差距。
💡 要点总结:
3D-score 的价值在于:
提升可信度:不只是能量最低的就算“最好”,而是把“重复出现 + 能量合理”的结合模式优先挑出来。
减少误判:避免错误构象“钻空子”得到较低能量而被误选。
一句话总结:3D-score 给了我们一把更稳妥的尺子,让对接结果不再靠运气挑选。
五、受体构象对预测的限制
即使算法再强,如果用的蛋白质结构本身不合适,预测依然会失准。为什么?因为
糖分子结合往往伴随蛋白质残基的局部调整。
作者把不同蛋白–糖体系分为三类情况(见 Figure 5A):
- Type 1:构象稳定型
结合位点残基在 apo、holo 和 AlphaFold3 模型中差别很小(RMSD < 2 Å)。
在这种情况下,WIIGA 表现稳定,能准确复现结合模式。
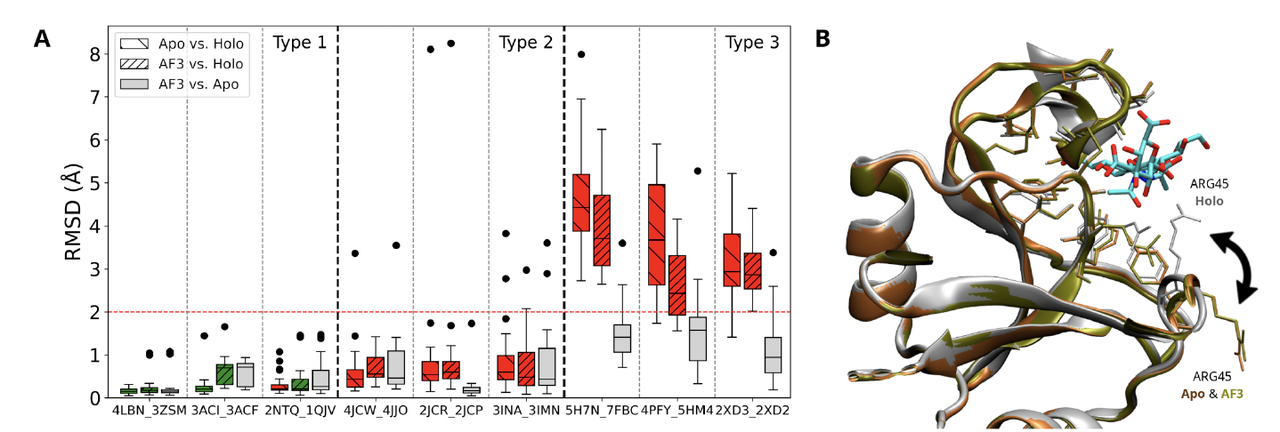
- Type 2:局部偏差型
大部分残基没问题,但关键残基有较大位移。
例如:在 murine CD44 的例子里,ARG45 偏移导致失去了与糖分子的关键氢键。结果是:算法再怎么引导,也预测不准(Figure 5B)。
- Type 3:全局偏差型
大部分结合位点残基都有显著差异(RMSD > 2 Å)。
这种情况下,WIIGA 的表现大幅下降,预测的姿势与真实结构偏差明显。
💡 要点总结:
算法不是万能的,如果蛋白质结构本身与真实结合态差别太大,预测必然受限。
关键残基的小偏差就可能导致大失败。
因此,在糖分子对接中,选择合适的受体构象(最好是 holo,或经过实验数据修正)非常关键
六、拓展应用:药物型配体的跨对接
除了天然糖分子,研究者还经常设计 药物型糖模拟物(glycomimetics) 来模仿糖的结合方式,以达到抑制或调控蛋白功能的目的。问题是:这些模拟物结构多样,如果对接方法不够精准,很难预测它们的真实结合模式。
作者测试了 WIIGA 在两个体系上的表现:
Galectin-3
β-1,3-Glucanosyltransferase
📊 对应 Figure 6
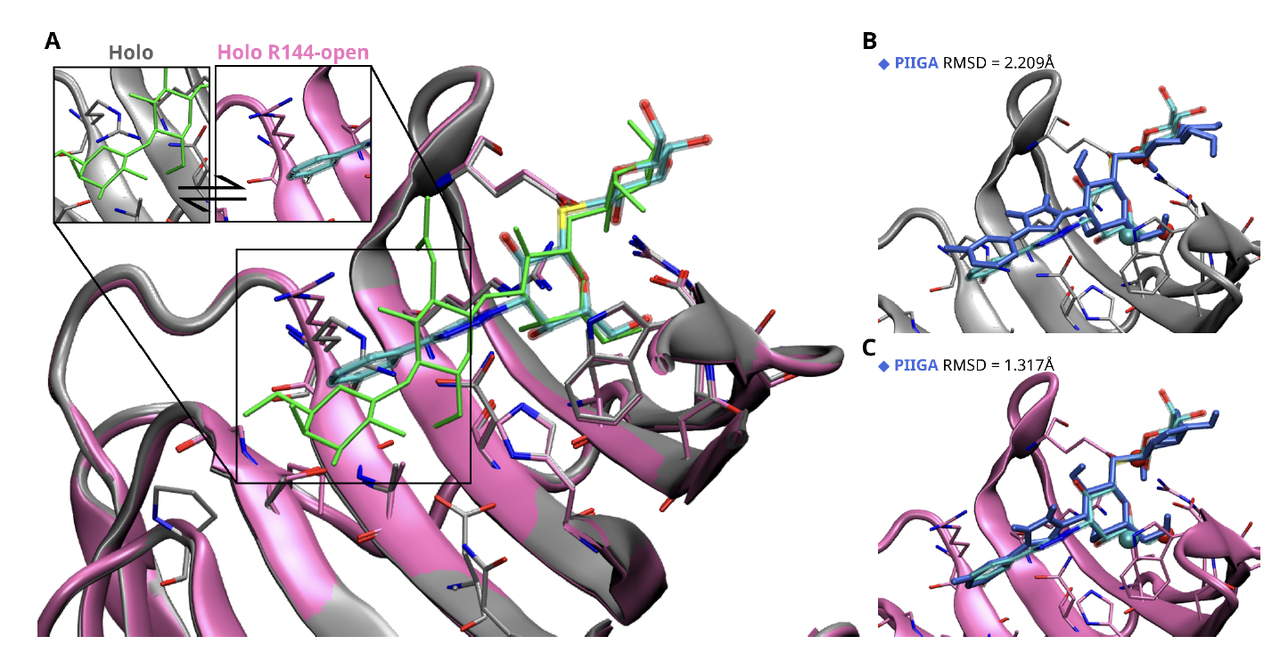
Panel A:展示了 Galectin-3 的两种受体构象(闭合型 R144-Closed 与开放型 R144-Open)。差别只在一个关键残基(ARG144)的摆放,但这就决定了预测结果是否可靠。
Panel B:在闭合型构象下,用 PIIGA 协议预测 KOW 配体时,结果出现了明显的空间冲突,说明这种受体构象不适合预测。
Panel C:在开放型构象下,预测结果与真实结合高度吻合,说明 合适的构象 + 引导对接 才能得到准确结果。
在 β-1,3-Glucanosyltransferase 的体系中,WIIGA 和 PIIGA 一致成功预测了药物型配体的结合方式,尤其是糖基部分几乎完全贴合原始配体,只在非糖部分有轻微偏差。
💡 要点总结:
WIIGA 不仅适用于天然糖分子,还能推广到药物型糖模拟物。
这意味着它能直接服务于 药物发现,帮助研究者筛选和优化糖基药物设计。
不过前提依然是:受体构象要选对,否则算法再强也无济于事。
七、总结与前景
这篇工作针对结构糖生物学中的老大难问题——如何准确预测蛋白–糖分子的结合模式,提出了一个新思路:利用结晶水分子的信息作为“历史线索”,引导对接。
整篇文章的亮点可以归纳为:
- 创新性方法
提出了 WIIGA 协议,把结晶水 → 理想相互作用点 → 对接偏好,形成完整流程。
配合 3D-score,大幅提升了正确姿势的识别率和可信度。
系统验证
在 30 个蛋白–寡糖复合物数据集上,WIIGA 成功率明显超过现有方法(53% vs. 30–43%)。
不仅能预测天然糖,还能扩展到 药物型糖模拟物,展示了药物设计潜力。
- 关键发现
受体构象是限制预测精度的主要因素,尤其是关键残基的小偏差就可能让结果失真。
因此,未来的预测应结合 多构象模型或实验数据修正。
- 应用前景
在基础研究上,WIIGA 为解析糖分子识别提供了新工具。
在应用层面,它有望助力 糖基药物设计,提高对药物型配体的预测准确度。
蓝极总结:
WIIGA 就像在蛋白质结合位点里安装了“导航标志”,让糖分子能更快更准地找到正确的结合方式,为基础研究和药物开发都打开了新可能。
延伸阅读
本文属于 AI4S文献 栏目。
返回 AI4S文献 → 去公众号阅读完整版 →