入门必读|当我们在聊分子动力学模拟时,我们在聊什么?
蓝极说: 如果你没有什么基础又想了解分子动力学模拟,看这篇就对了!
背景
想象一部惊悚片:主角凭借“完美”的监控一路追踪反派,踹开仓库门,却发现空无一人——目标早在几小时前就消失了。我们用对接算法在一个下午把数百万个“完美”结合体排了名,结果也几乎如此。湿实验的结论?大多是海市蜃楼——纸面上耀眼,真正验证时却像哑炮。

正如静态监控抓不住真实运动,分子对接只给出漂亮的静态快照,却错过了分子生命的动态现实。计算预测与实验事实之间的这道鸿沟,正是基于结构的药物发现(structure-based drug discovery, SBDD)所面临的核心挑战。对接是高效的起点,但终究只是快照:蛋白质被近似为基本刚性,水环境被高度简化,而熵效应则在很大程度上被忽略。
超越静态快照
蛋白质并不是凝固的雕塑;在溶液中它们会不断弯曲、摆动,可能暴露出隐藏的口袋,也可能让看似存在的口袋塌陷。分子对接无法捕捉这种运动,更无法计算将一个柔性的配体“锁定”时所付出的熵代价——这往往就是“完美”结合体在实验中失败的原因。此时,分子动力学(Molecular Dynamics, MD)模拟登场:它让蛋白质和配体在纳秒到微秒的尺度上真实运动,从而帮助我们判断某个构象是否真正稳定。当 MD 与自由能微扰(Free Energy Perturbation, FEP)等方法结合时,能给出的结合亲和力预测比单纯的对接更接近现实。
对接的局限性并不只是理论上的挑刺。大型基准测试,如 D3R Grand Challenge,一次又一次地揭示了对接在实践中的困境。即便某个配体的结合姿势与晶体结构的偏差小于 2 Å,看上去很合理,但评分函数依然常常误判其真正的结合强度。原因在于:受体柔性、溶剂效应以及熵惩罚被过度简化,甚至完全忽略,导致对接得分与实验亲和力渐行渐远。
结果就是:姿势看似合理,但预测往往失准。
在计算机中模拟生命
对接(docking)是一张快照,而分子动力学(MD)是一部电影。在 MD 中,计算机不只是把形状对齐然后判定“契合”,而是要真正为体系中的每一个原子——包括蛋白质、配体以及周围的水分子——逐步求解牛顿运动方程。结果就是一部逐帧展开的“分子电影”,展示体系随时间的演化:侧链在摆动,水分子进出结合口袋,口袋有时张开得更大,有时又瞬间闭合。与其说是死板的“锁钥模型”猜测,不如说 MD 呈现的是分子相互作用的真实面貌:动态、柔性、持续变化。
这种方法的价值主要体现在两个方面:
构象优化(Refinement):它能告诉你,一个对接构象在蛋白质构象发生变化后是否依旧稳定,还是会迅速解体。
热力学信息(Thermodynamics):通过轨迹,可以估算结合自由能,即配体留在口袋里与游离漂走之间,哪种状态更有利。
当然,这些计算也极其繁琐。为了捕捉有意义的运动,尤其是在高度柔性的靶点中,往往需要数百纳秒甚至微秒级的长时间模拟,耗费大量计算资源。
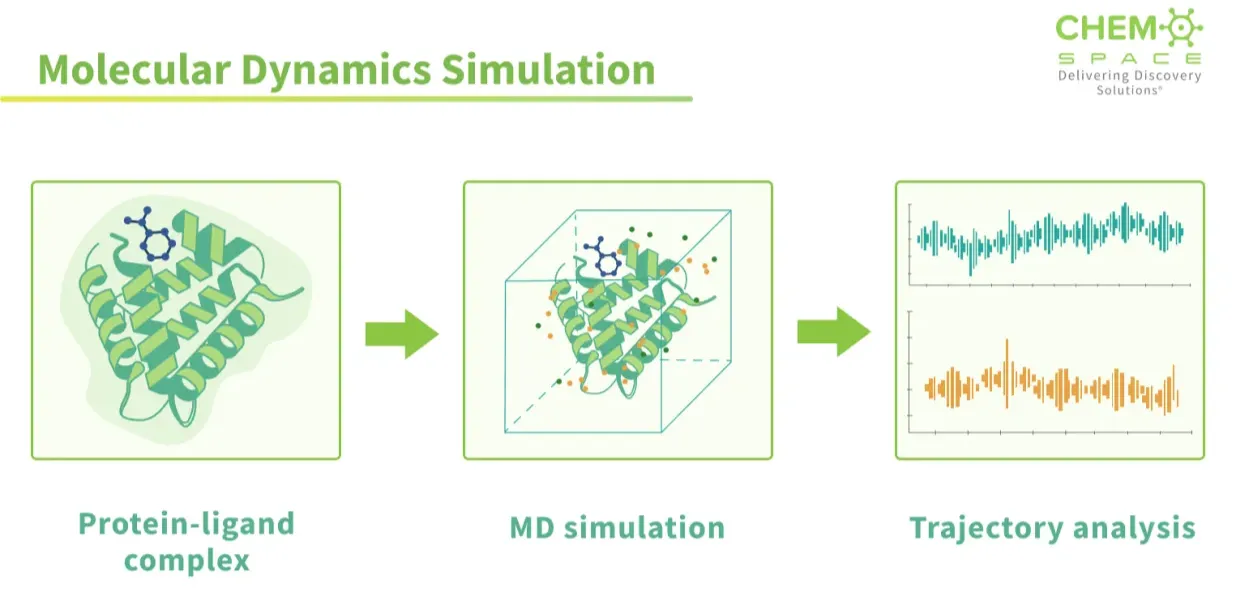
力场及其重要性
如果把分子动力学模拟比作一部电影,那么力场(force field)就是剧本。原子本身并不懂物理,计算机需要一整套规则来定义:化学键可以被拉伸到什么程度、键角会如何弯曲、电荷之间如何相互吸引,以及范德华力如何相互推拉。简而言之,力场就是一组方程和参数,把牛顿定律转化为模拟可以使用的数字化语言。
不同的 MD 软件在实现这些规则时会有所差异。有些更注重生物分子的精确性,有些强调计算速度和 GPU 加速性能,还有一些则突出灵活性,方便研究者进行定制化设置。下表展示了几种常用的 MD 引擎,以及它们在力场和相关功能上的特点:



增强采样与结合自由能的作用
分子动力学模拟的确能带来“动态电影”,但它的节奏往往慢得令人抓狂。大多数模拟只能跑到微秒级,而真正的生物学过程却常常发生在毫秒甚至秒的尺度上。这就像你想看一部 2 小时的电影,却只能截取几秒钟的片段,关键情节自然全部错过。
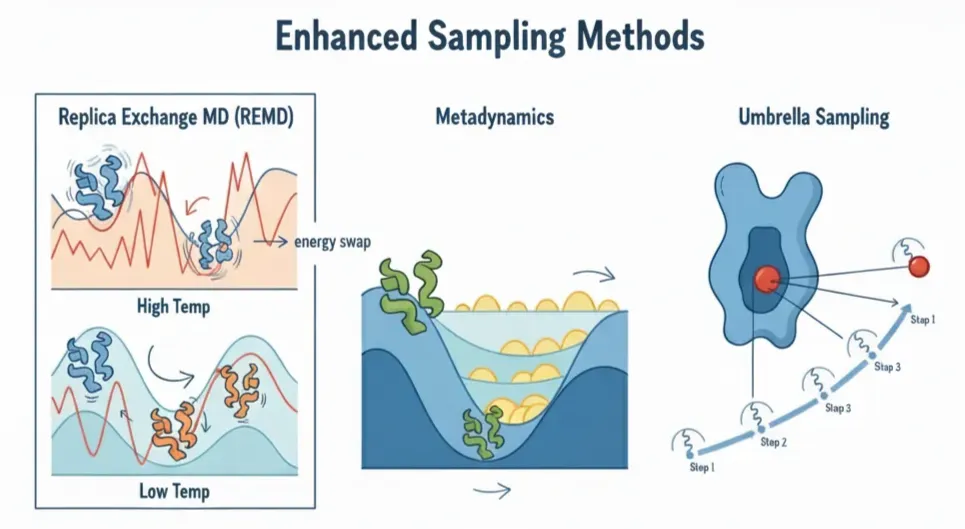
这时,增强采样方法(Enhanced Sampling Methods)登场了:它们会在规则上做些“微调”,让我们能够快进跳过那些冗长无聊的情节,直接捕捉稀有而关键的高价值片段。就像电影没有一个统一的“跳过无聊剧情”的按钮一样,MD 也不存在单一的加速方法。常见的增强采样策略包括:
Replica Exchange MD (REMD):在常规 MD 中,体系往往被困在局部能谷,因为在室温下它没有足够能量翻越势垒。REMD 的做法是运行一组在不同温度下的“克隆”体系。高温副本震动更剧烈,更容易越过能垒,然后再与低温副本交换。结果是:低温体系也能接触到那些原本无法企及的高能态。
Metadynamics:蛋白质喜欢“赖”在同一个舒适的能谷里。Metadynamics 的策略是不断往体系已经访问过的状态里加上小的“能量丘”(高斯函数),阻止它反复回到原处,把能量地形逐渐“填平”,从而迫使体系探索新的构象。
Umbrella Sampling:有些过程(例如把配体从蛋白质口袋里拉出来)能量障碍太陡峭,直接模拟时配体会立刻掉回去。Umbrella Sampling 把整个过程分解成一系列小步骤,每一步都用轻柔的限制力(像弹簧一样)稳定住,让体系逐段完成过渡。
其他技巧(ABF、Funnel MD 等):这些方法更为专业,但核心思路相同:在体系中加入适度的偏置帮助它跨越能垒,再在后续分析中小心地去除偏置,从而恢复出真实的物理结果。
当我们已经探索过不同状态,并确认配体确实能够稳定结合时,下一个关键问题就是:“这种结合到底有多强?”
这正是结合自由能(ΔG_bind)的意义所在。它是一个定量指标,用来衡量配体与蛋白质结合的紧密程度,并且可以直接与实验测得的解离常数(Kd)对应。
然而,计算结合自由能并不容易,因为它既要捕捉焓(相互作用能量),也要考虑熵(分子结合时失去的自由度)。多年来,计算化学家们构建了一整套方法论工具箱,在精度与速度之间做取舍:
Docking scores:速度极快,非常适合在海量分子库中做初筛,但并不是真正的自由能。
MM-PBSA(分子力学–泊松-玻尔兹曼表面积法):利用 MD 轨迹的快照,分别计算结合态与游离态的能量,并加入隐式溶剂效应。方法便宜,趋势有时能对,但绝对值误差往往非常大。
LIE/ELIE(线性相互作用能):通过提取配体与蛋白之间的主要相互作用(静电 + 范德华力),再结合少量从实验数据拟合的系数,来估算结合自由能。速度快,效果也算合理,但准确性依赖训练数据。
FEP(自由能微扰):自由能计算的“重量级冠军”。它通过一系列模拟,逐步将一个配体“炼金术般”地转化为另一个配体(无论是在蛋白口袋里还是在溶液中),从而得到结合自由能的差异。如果操作得当,其精度可以接近实验结果。代价是:需要极高的算力投入,同时要求使用者具备丰富经验。


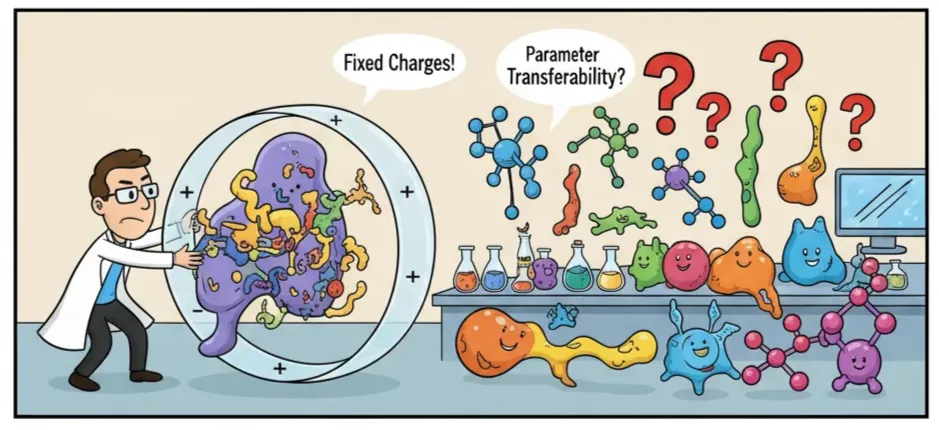
经典力场的局限
几十年来,分子模拟一直依赖于 AMBER、CHARMM、OPLS 等经典力场。它们就像科研里的“老黄牛”,支撑了无数发现与药物设计项目。但这些力场并非完美无缺——它们的简化假设总要付出代价。
最大的问题在于:电荷固定。真实的分子并不是静态的,它们的电子云会不断移动、极化,甚至在某些情况下会发生电荷转移。而经典力场却把电荷“钉死”在原位。这让它们在带电体系或高度极性的环境中精度不足。
另一个问题是:参数的可迁移性差。一个力场可能在某类分子上表现良好,但换成另一类分子就完全失效。研究人员往往不得不重新进行繁琐的参数化工作,才能让模拟跑得起来。
神经网络力场:新的前沿
这正是机器学习发挥作用的地方。神经网络力场并不依赖于固定的方程,而是直接从高水平的量子力学(QM)数据中学习能量面,这些计算能够真实捕捉电子的行为。正因为直接学习自 QM,这类模型能感知到经典模型无法触及的效应。例如:
极化效应:在现实中,当另一个电荷靠近时,原子周围的电子云会发生畸变。经典力场把电荷“冻住”,但神经网络模型能够动态响应。
电荷转移:有时电子会从一个分子转移到另一个分子(在结合口袋中很常见)。经典模型无法处理,而神经网络可以。
多体效应:经典力场往往将相互作用拆分成一对一(原子 A 拉原子 B),而神经网络能捕捉多个原子同时作用的集体影响。
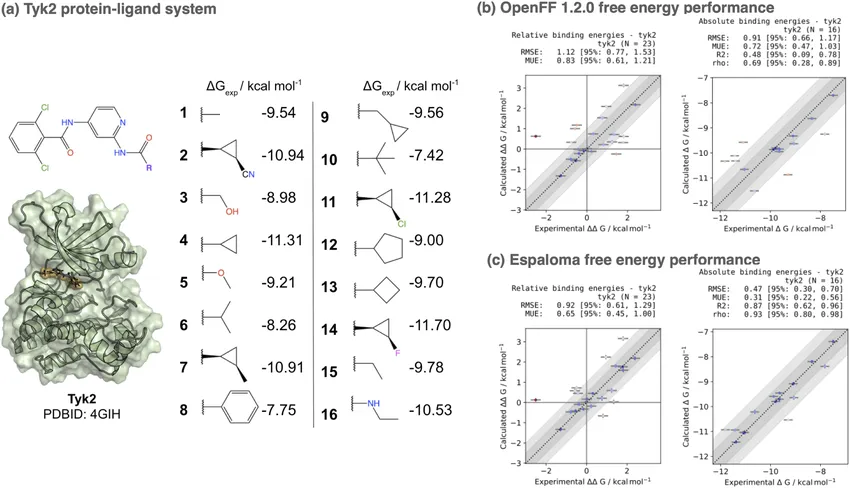
这种差异并不只是理论上的。在一个基准测试中,神经网络力场 Espaloma 在 Tyk2 激酶体系上的表现优于常用的经典力场 OpenFF 1.2.0,把预测误差从 1.10 kcal/mol 降低到 0.73 kcal/mol。在药物发现中,哪怕是几分之一 kcal/mol 的差异,都可能彻底改变候选分子的排序,这无疑是一个重大突破 [3]。
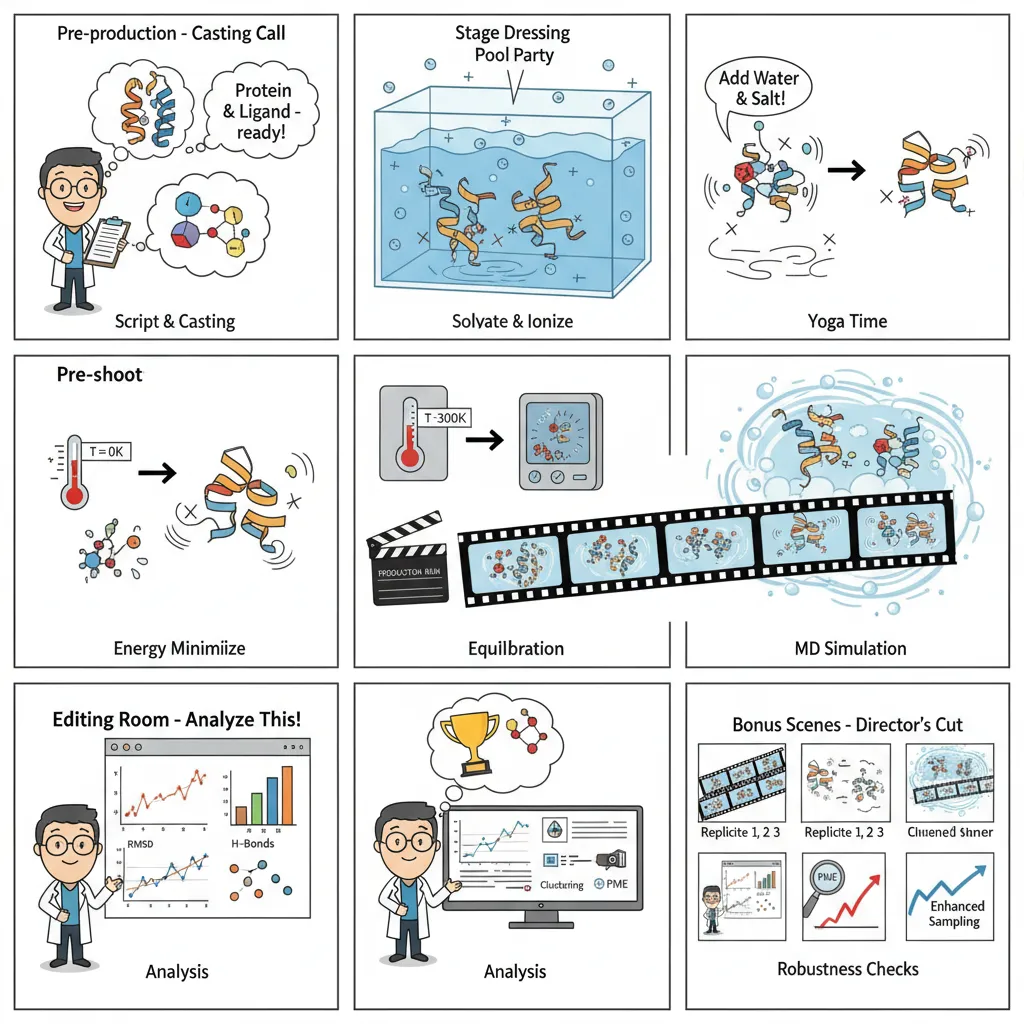
一场分子动力学模拟究竟如何运行?
可以把分子动力学(MD)想象成搭建一个电影制片厂:先用物理定律布置舞台,再去捕捉原子间的动态舞蹈。
- 体系准备:剧本与演员阵容
首先要准备好蛋白质和配体,修复缺失的原子,分配电荷,并选择合适的力场。如果这一步做得不好,就像主演选错了人——整场戏都会垮掉。
- 环境创建:搭建舞台
分子不会生活在真空里。我们把它们放进一个水盒中,加入离子,并设置符合生理条件的盐浓度,从而让分子间相互作用更接近真实的生物环境。
- 能量最小化:安全检查
开始“开机”前,需要先放松体系中的紧张部位。这个步骤能消除不现实的高能量位置,否则模拟可能因为糟糕的初始构象而“崩溃”。
- 平衡阶段:彩排
逐渐把体系加热到体温(300 K),同时保持主体结构稳定,让水分子和侧链找到自然的运动节奏。这样能确保体系在正式运行前处于稳定状态。
- 生产运行:正式拍摄
这是模拟的核心阶段。约束被解除,体系自由演化,我们在纳秒到微秒的时间尺度上记录原子的运动轨迹。这些快照最终拼接成我们的“分子电影”。
- 结果分析:剪辑室
电影拍完之后,就要逐帧解析:追踪 RMSD、氢键、构象变化,或者计算结合自由能。在这里,原始的运动数据转化为科学洞见。
- 验证与增强:质量控制
最后,需要通过重复模拟来检查结果是否一致,确保统计学上的可靠性。如果有必要,还会引入增强采样方法,去捕捉那些稀有但关键的事件。
MD 依然难以逾越的壁垒
分子动力学(MD)能提供极其精细的原子级细节,但它并不是万能的。原因有很多,例如:
- 时间尺度问题
模拟通常只能跑到纳秒至微秒,而生物过程发生在毫秒乃至秒的范围。这就像你想理解一首交响乐,却只听到每个音符的几微秒。增强采样能帮上一些忙,但我们依然与真实生命节奏相差几个数量级。
- 精度上限
即使是最先进的自由能方法,精度也往往止步在约 0.5–1.0 kcal/mol。听起来微不足道,但在结合亲和力上,这意味着 3–5 倍的误差。在药物研发中,纳摩尔和微摩尔的差别足以决定成败,这样的误差足以彻底改写候选分子的优先级。
- 可迁移性差距
没有哪一种力场或神经网络模型能“通吃一切”。在可溶性蛋白上表现出色的模型,放到膜蛋白上可能就完全失效;在激酶上训练得很好的方法,遇到 GPCR 就可能跌跟头。没有通用的配方,每一个新靶点都需要痛苦的重新验证。
- 采样的幻觉
即使采用了增强采样方法,也没人能保证“看遍一切”。某个隐匿的变构口袋可能要到 10 毫秒尺度才会打开,而你的微秒模拟永远捕捉不到。错过这些稀有状态,就可能错过关键的耐药机制或选择性机制。
- 计算的现实考验
没错,AI 力场和 FEP 可以接近实验精度,但它们的计算需求惊人,并且需要专业技能。学术实验室和小型生物技术公司往往夹在昂贵的“严谨方法”和便宜但靠不住的近似方法(如 MM-PBSA)之间。
- 验证的瓶颈
真正的瓶颈不在硅片,而在生物学。你可以在一周内跑完上千个 MD 任务,但在实验室里验证十个化合物可能要耗费数月。
结论
分子动力学并不仅仅是对接(docking)的“附加组件”,而是连接静态预测与生物现实的桥梁。通过在计算机中让蛋白质和配体“呼吸”,MD 能帮助我们区分真正的结合体与假阳性,优化对接构象,并首次揭示结合所需付出的热力学代价。它无法取代实验,但却能极大缩小搜索空间,为湿实验团队提供更高质量的假设。
MD 的未来在于物理与机器学习的交汇。神经网络力场、GPU 加速以及更高效的增强采样策略,正在推动模拟逐步接近生物学的时间尺度,并接近实验精度。结合智能化的筛选流程(docking、FEP、AI 驱动的构象打分),MD 将继续是把计算预测转化为可操作候选分子的最有价值工具之一。目标不仅是“再现”现实,而是去引导现实:帮助优先选择正确的分子,减少无效合成,加速从想法到药物的转化之路。
参考文献:
[1] D3R Grand Challenge Overview – Community-wide evaluation of computational drug design methods.
[2] Chemspace – Molecular Dynamics Simulations: Concepts and Applications
延伸阅读
本文属于 AI4S文献 栏目。
返回 AI4S文献 → 去公众号阅读完整版 →