“人类互作组”的开山之作:Science揭示最大规模蛋白互作预测
蓝极说:
我愿称这篇文章为“人类互作组”的开山之作!
第一部分:背景介绍
蛋白质之间如何相互作用,决定了几乎所有细胞生命活动的进行。从信号转导、能量代谢,到免疫防御、疾病发生,蛋白质-蛋白质相互作用(Protein-Protein Interactions, PPI)都扮演着核心角色。
然而,我们对人类蛋白质互作网络的认知仍然远远不够。实验方法,如酵母双杂交(Y2H)和亲和纯化-质谱(APMS),虽然在过去几十年推动了互作组学的发展,但它们往往存在两个问题:
假阳性与假阴性率高,导致结果不够可靠;
在非生理条件下测得的互作,可能无法反映真实的体内情况。
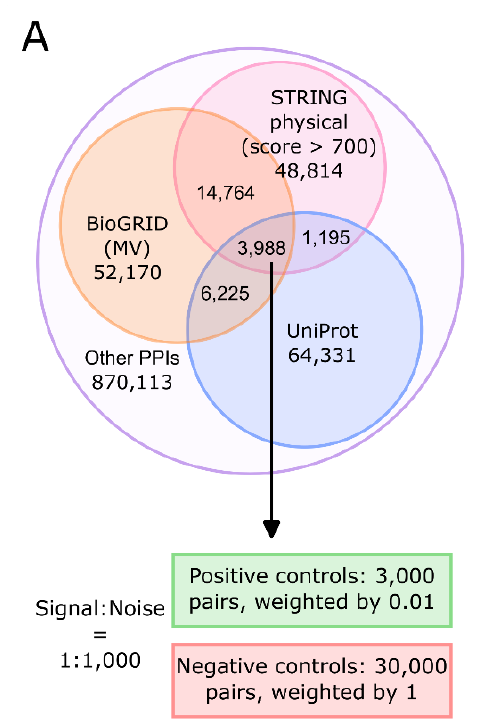
一个直观的例子就是文章中的 图1A:研究团队对比了三大主流数据库(UniProt、BioGRID、STRING)的“高置信度”互作数据,结果发现三者的交集仅有3988对。这意味着,即便是我们最“可信”的实验结果,不同数据库之间的重合度也极低,显示出目前人类互作图谱的碎片化和不一致性。
正因如此,人类完整的“互作组地图”一直被视作系统生物学的重大挑战。近期,来自德州大学西南医学中心的丛倩教授(Qian Cong)团队联合华盛顿大学David Baker实验室与首尔大学Minkyung Baek团队,在 Science 杂志(2025年9月)发表了重要研究成果:
“Predicting protein-protein interactions in the human proteome”(Zhang et al., Science 2025, DOI: 10.1126/science.adt1630)

这项工作首次在人体水平上,通过大规模计算预测,构建出了迄今为止最大、最精确的人类蛋白质互作图谱。这不仅极大地扩展了我们对细胞内蛋白质网络的理解,还为疾病机理研究和新药靶点发现提供了前所未有的资源。
第二部分:技术突破——从30 PB数据到新一代深度学习网络
面对“人类互作组预测”的巨大挑战,作者团队在数据和算法两个层面上都做出了突破性的创新。
- 数据层面的革新:omicMSA
在蛋白质互作预测中,一个核心依赖就是共进化信号。如果两个蛋白在进化过程中总是协同变化,那么它们更可能存在互作关系。但要捕捉到这样的信号,前提是 足够深的多序列比对(MSA)。
过去的互作预测主要依赖 UniRef 等数据库构建MSA。然而,这类数据库虽然整理得很规范,但覆盖面有限,尤其是在多细胞真核生物中,序列数量不足,导致MSA往往太“浅”,无法提供稳定的共进化信息。
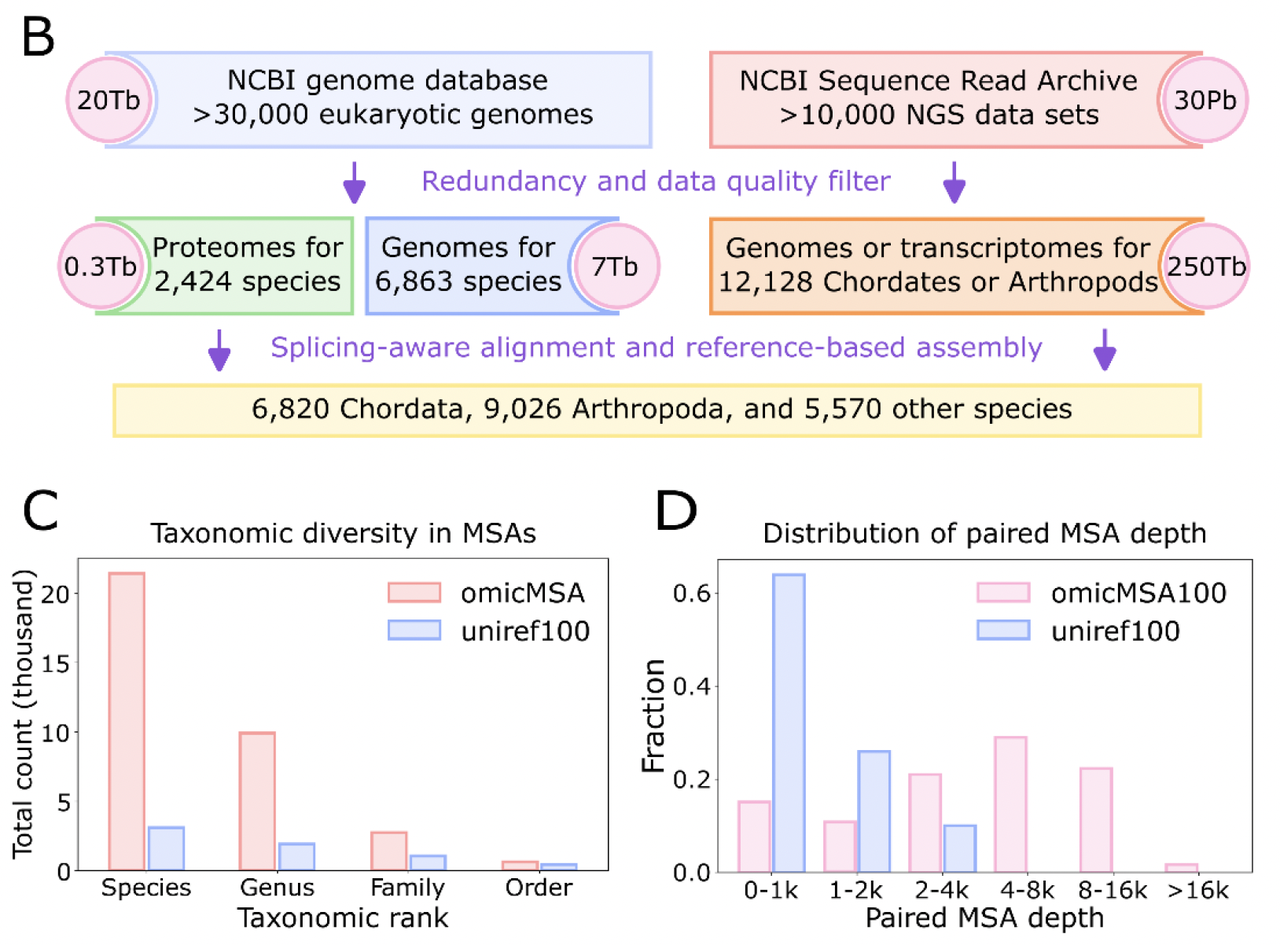
这篇文章的作者采取了一条更“硬解”的路线:直接从 NCBI的基因组和转录组数据 中构建MSA。
他们收集了 21,414个真核物种 的数据,数据量超过 30 PB。
其中只有约20%的物种有完整的蛋白注释,大部分需要研究团队自己完成组装与蛋白预测。
最终得到的MSA被称为 omicMSA,它的平均深度相比UniRef提高了 7倍。
这种大规模数据处理带来的提升非常直观。
Fig. 1B–C 清楚地展示了omicMSA的构建流程,以及与传统数据库相比在覆盖度上的差距。
更深的MSA意味着更丰富、更可靠的共进化约束信号,直接提升了互作预测的灵敏度和稳定性。
Fig. 2D 的结果进一步说明了这一点:在相同的模型框架下,使用omicMSA比使用colabMSA能显著提高预测的召回率。
omicMSA提供了一个新的“数据底座”。它不仅弥补了公共数据库在真核生物中的不足,还让深度学习模型第一次能够在全人类蛋白质范围内,依赖真正足够的进化信息来进行互作预测。
omicMSA的出现,相当于把人类互作预测的输入数据质量提升了一个量级,这是整篇文章能成立的前提条件之一。
- 算法层面的革新:RF2-PPI
如果说 omicMSA 解决了“输入数据不够深”的问题,那么 RF2-PPI 就是专门为“如何更快、更准地预测蛋白互作”而设计的新工具。
为什么需要新算法?
现有的结构预测工具(例如AlphaFold-Multimer)在处理蛋白互作时有两个局限:
速度不足 —— AlphaFold设计的初衷是单体或少量复合物建模,计算一个复合物往往需要几分钟到几个小时。面对 1.9亿蛋白对,几乎不可行。
缺乏“否定能力” —— AlphaFold总是会给出一个复合物结构,但不擅长判断“一对蛋白是否真的互作”。这就导致它在大规模筛选时会引入大量假阳性。
因此,需要一种既能快速处理大规模数据,又能明确区分“真互作”和“假互作”的新模型。
RF2-PPI的核心思路
RF2-PPI的设计基于RoseTTAFold2的架构,但加入了几个关键改进:
训练数据的扩展:
传统方法依赖PDB里的复合物结构,但数据量有限。作者在此基础上,又利用了 2亿个AlphaFold单体模型,从中切分出结构域(domain),并筛选得到 22万+ 结构域-结构域互作(DDIs)。
这让训练集规模比PDB里的真实互作大了 17倍,极大缓解了数据不足的问题。
输入信息的优化:
- RF2-PPI不仅使用单体的MSA,还使用“配对MSA”(paired MSA),结合omicMSA的数据优势,能更好地捕捉共进化信号。
目标函数的改造:
- 模型训练时引入了“正/负样本对比”机制:即不仅学习已知互作的特征,还要学习如何拒绝随机配对的蛋白。这一点正是AlphaFold所欠缺的。
性能与效率
性能:在 Fig. 2D 中,RF2-PPI结合omicMSA,在高精度要求(80%–90% precision)下的召回率明显超过ColabFold和AlphaFold,尤其是在小界面、弱互作上表现更好。
效率:RF2-PPI比AlphaFold快约 20倍。这意味着过去不可想象的全人类规模预测,在这套框架下真正变得可行。

RF2-PPI的价值在于:它把“预测蛋白是否互作”从AlphaFold的副产品,变成了一种专门优化过的能力。速度和准确性的提升,使得人类互作组的全景预测第一次真正落地。
数据深度 + 算法创新 的双重突破,使得这项工作第一次能够在“人类蛋白质全景图”层面上,既规模庞大,又保持高置信度地预测PPI。
第三部分:成果——规模与精度的双重突破
在解决了数据和算法的瓶颈之后,研究团队将这套框架应用到全人类蛋白质组,得到了目前最大规模的蛋白质互作预测成果。
- 从1.9亿对到数万高置信度互作
研究起始于 19,528个人类蛋白(AlphaFold数据库中有结构的条目),所有可能的组合约 1.91亿对。
首先利用 DCA(直接耦合分析) 进行粗筛,去掉明显不相关的组合。
接着用 RF2-PPI 对候选互作进行预测,进一步缩小范围。
最后再用 AlphaFold2 精细建模,对最有可能互作的蛋白对进行结构层面的验证。
整个流程层层筛选,既保证了规模覆盖,又确保了预测质量。
- 结果:17,849对高置信度互作
最终得到的结果:
29,257对互作 在80%精度阈值下成立;
其中 17,849对 互作达到90%精度,被视为“高置信度”。
更重要的是,其中有 3,631对互作此前没有任何实验数据支持,属于真正的“新增发现”。
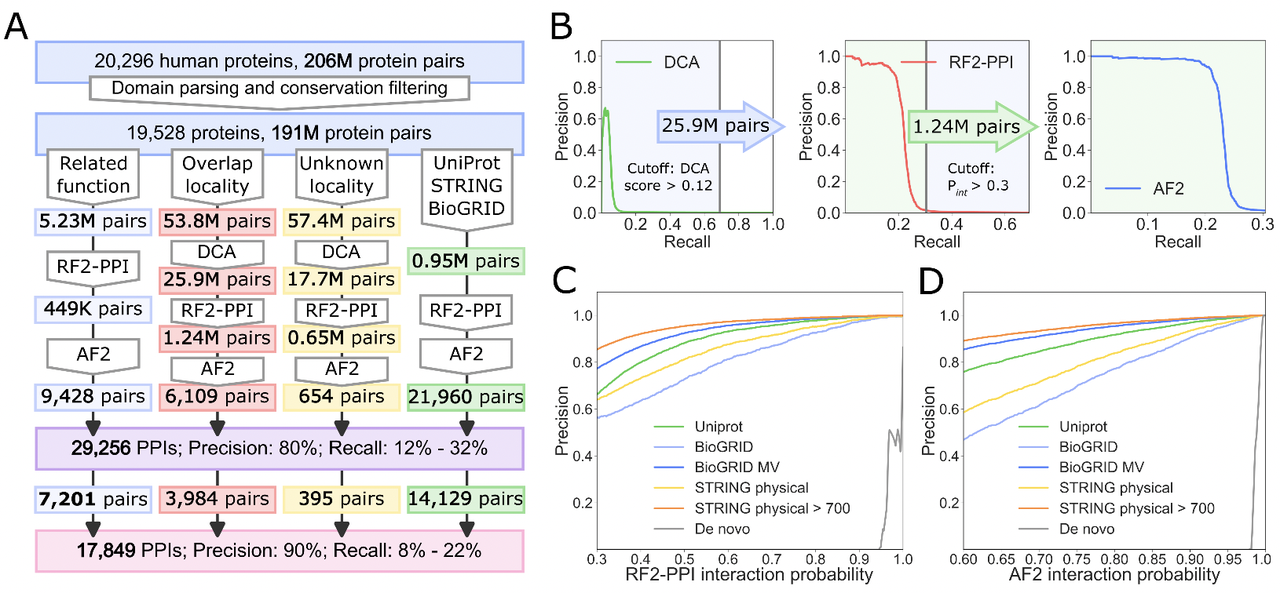
Fig. 3A:展示了整个筛选流程,从1.9亿对一路缩小到最后的数万对。
Fig. 3B–D:性能评估,证明RF2-PPI + omicMSA显著提高了准确率。
- 可靠性:功能与定位的一致性
为了验证这些预测的可信度,作者进一步检验了互作蛋白的生物学属性:
在功能注释(Gene Ontology, GO terms)上,互作的蛋白往往属于同一类生物学过程;
在亚细胞定位上,预测的互作蛋白也倾向于出现在相同的细胞区室。
这种功能/定位的一致性是互作真实性的独立佐证,也说明这些预测并不是“模型幻觉”。
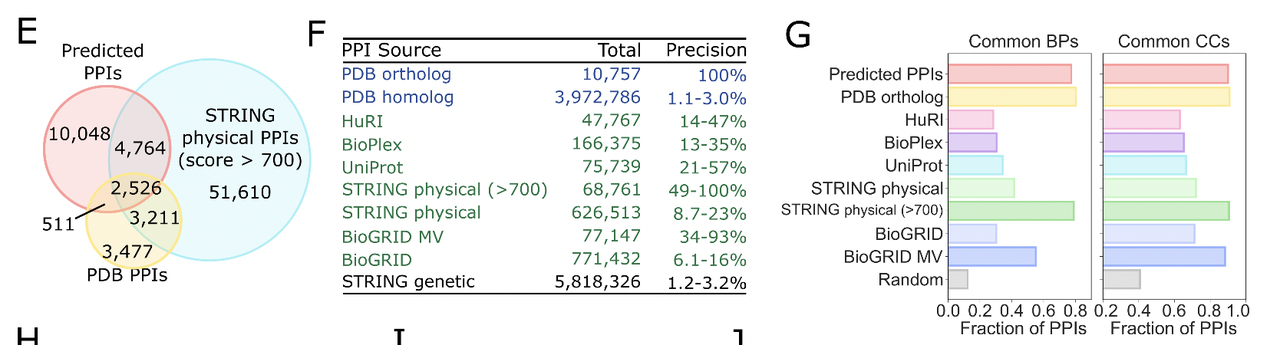
Fig. 3F–G:显示预测互作显著富集在功能一致和定位一致的组合中,而随机配对则没有这种趋势。
- 扩展已有的三维互作结构库
除了互作对本身,这些预测也带来了高质量复合物结构的“爆炸式扩展”。
已知高分辨率复合物数量在几千个量级;
本文的预测新增了数万个结构模型,使得三维互作组的规模扩展了 近三倍。
这意味着,现在研究者能够以前所未有的规模,从结构层面理解蛋白质网络。
这项工作交付了迄今为止规模最大、精度最高的人类蛋白质互作图谱,既补全了已有知识的空白,也为发现全新的生物学规律提供了资源。
第四部分:生物学与疾病机制的新发现
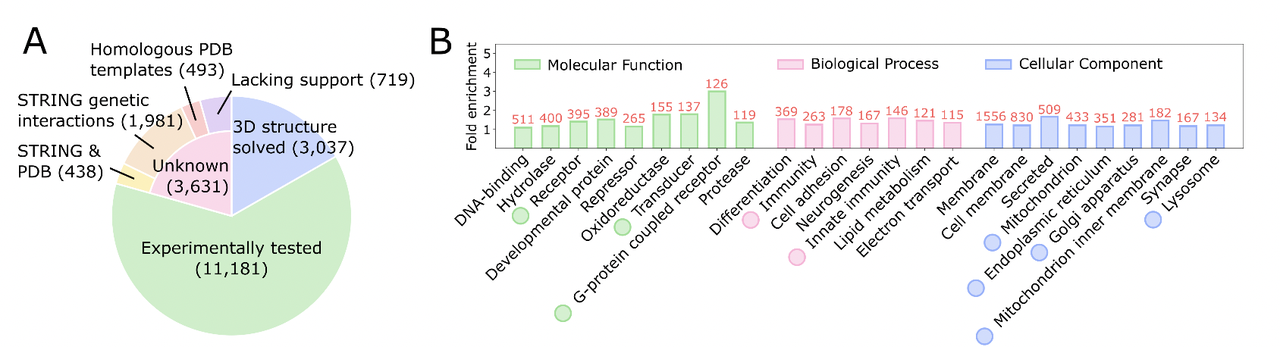
除了规模上的突破,这套预测真正的价值在于:它能揭示新的生物学机制,尤其是和疾病相关的互作线索。 作者团队挑选了几个典型案例,展示这些预测如何帮助我们理解生命过程。
- GPCR信号通路的新互作(Fig. 4C–D)
GPR143 – HPS1:
- GPR143是一种定位在溶酶体和黑素体的GPCR,已知与眼部白化病相关。预测显示它与HPS1(一种参与黑素体形成的蛋白)直接互作。更有意思的是,GPR143的多处致病突变正好落在预测的互作界面上,这强烈暗示互作破坏可能是疾病机制之一。
C5AR2 – ELANE:
- C5AR2是免疫细胞表面的炎症受体,而ELANE是中性粒细胞的蛋白酶。预测结果表明二者互作,可能是一种负向调控:ELANE通过结合C5AR2抑制过度炎症反应。这为理解免疫平衡提供了新角度。
免疫调控中的互作新线索(Fig. 4E–F)
OTUD4 – ASB8:OTUD4是去泛素化酶,能稳定抗病毒蛋白MAVS;ASB8则是泛素连接酶家族成员。预测它们互作,提示可能存在一种互作网络:ASB8促进降解,OTUD4反向调控,二者共同维持先天免疫的平衡。
KLRG1 – TLR3:预测显示NK细胞抑制受体KLRG1可能直接结合TLR3(一个病毒RNA识别受体),这意味着抑制信号可能比我们以往理解的更“直接”,跨越了不同通路。
线粒体复合物的装配机制(Fig. 4G–H)
MT-CO2 – COX20:确认了已知的复合物IV组装因子作用。
C8orf82 – ETF复合物:一个此前功能未知的小蛋白C8orf82,被预测与电子转移黄素蛋白复合物结合。这暗示它可能是装配辅助因子或新的调控亚基。这样的发现为未注释蛋白的功能研究提供了重要切入点。
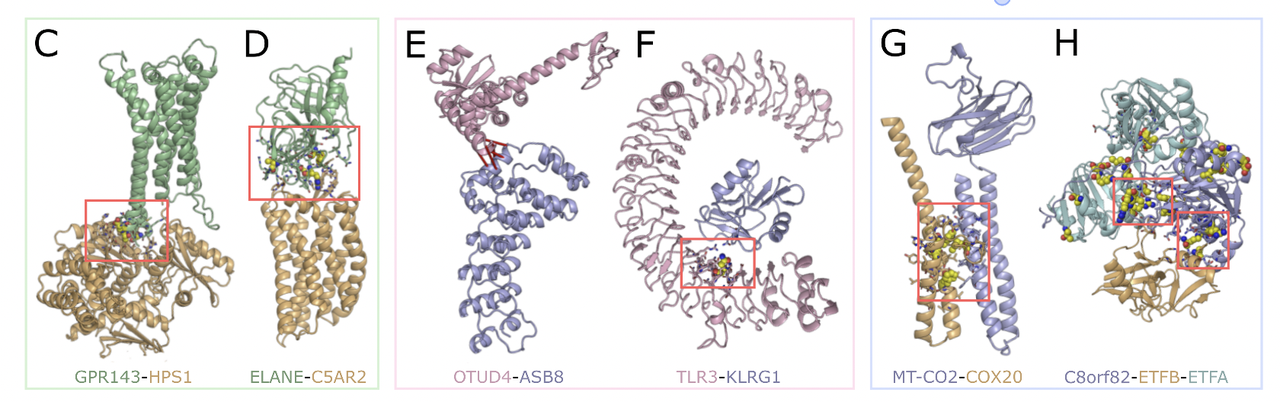
多蛋白复合物的新组成(Fig. 5)
作者根据预测结果,识别了 404个新的多蛋白复合物。
例如,微管相关的 Tubulin polyglutamylase(TPG)复合物 被预测新增了3个亚基(包括TBC1D19),而且这些新增亚基在结构上与已知催化亚基形成合理互作。
另一个例子是 NOL10、DDX47 等核相关蛋白,被预测形成一个新型复合物,可能同时参与转录调控与核糖体生成,揭示了两个核心细胞过程之间潜在的交叉点。
第五部分:意义与未来展望
这项工作不仅是一份“预测结果”,更像是人类互作组研究的一个新起点。
- 意义
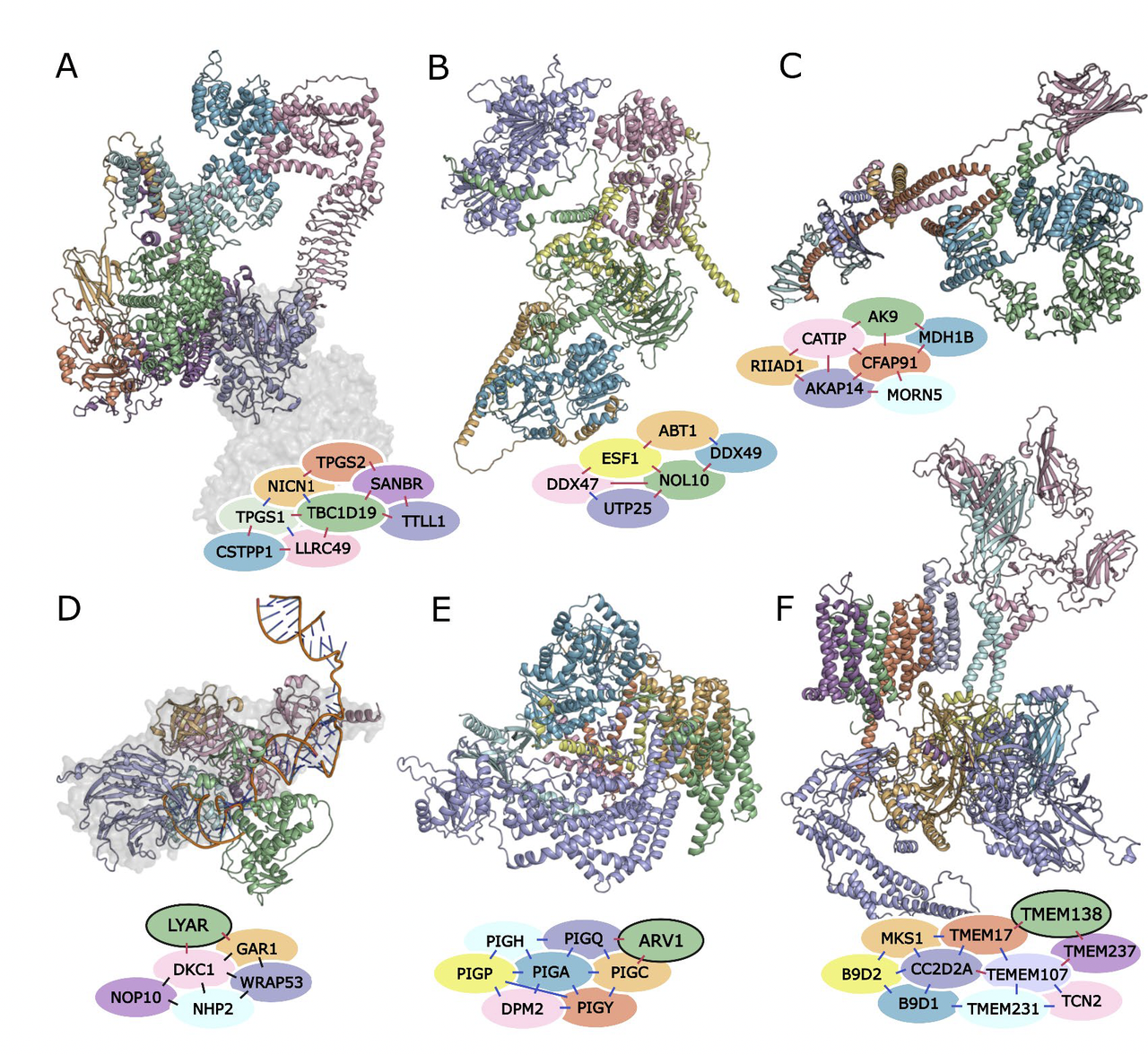
规模前所未有:这是目前最大的人类互作预测数据集,包含 17,849对高置信度互作,大约占人类互作组的 8–22%。
填补实验空白:超过 3,600对互作 过去从未被实验捕捉,尤其是膜蛋白、跨膜复合物等“实验盲区”。
疾病相关性强:作者将已知致病突变映射到预测互作界面,发现约 16,000个突变落在这些位置,说明PPI破坏可能是致病的重要机制。
功能注释的潜力:通过互作网络,许多未注释蛋白可以被推断出潜在作用路径,这是理解基因功能的重要途径。
- 局限
弱互作和瞬时互作难以捕捉:这些往往由无序区(IDR)介导,计算预测和实验检测都不擅长。
覆盖度仍有限:即便有近1.8万对高置信度预测,这可能只占全人类互作组的12–32%,距离完整图谱还有相当距离。
- 展望
数据会越来越深:随着更多物种的基因组测序,omicMSA将不断扩展,进化信号会更强。
方法会持续进化:未来的网络架构可能更好地处理IDR互作,弱互作的预测能力也有望提升。
实验与预测结合:计算预测可以提供大规模假设,实验方法则可以针对性验证,这种结合将加速人类**三维互作图谱(3D interactome)**的完整绘制。
这项工作为人类互作组研究提供了前所未有的资源和方法学突破,也让“完整的人类3D互作组图谱”从遥远的目标变成了可以预期的未来。
原文链接:https://doi.org/10.1126/science.adt1630
作者也开放了在线网站,大家感兴趣可以去在线网站进行互作预测。
网站链接:http://prodata.swmed.edu/humanPPI/
延伸阅读
本文属于 AI4S文献 栏目。
返回 AI4S文献 → 去公众号阅读完整版 →